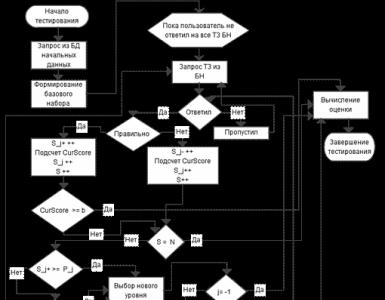
Anomalie delle vie urinarie superiori. Accademia medica statale di Omsk, Dipartimento di chirurgia pediatrica, malformazioni del tratto urinario superiore nei bambini, Dottore in scienze mediche. Pisklakov A.V. Il concetto di difetti congeniti ed ereditari
Anomalie renali: 1. Aplasia- assenza congenita di un rene. 2. Ipoplasia- riduzione congenita delle dimensioni dei reni. 3. Raddoppio- l'anomalia più comune. Con il raddoppio completo, ciascuna metà del rene ha un sistema pielocaliceale separato. Da ciascuna pelvi parte un uretere, che può aprirsi nella vescica con due orifizi o fondersi in uno solo a un certo livello. 4. Displasia renale- riduzione congenita delle dimensioni dei reni con alterato sviluppo del parenchima. 5. Accessorio, terzo rene si trova al di sotto del normale; ha un apporto sanguigno e un uretere indipendenti. 6. Distopia renale- anomalia della localizzazione: pelvica, iliaca, lombare, toracica. A differenza della nefroptosi, nella distopia i vasi del rene e dell'uretere sono corti. Si verifica la distopia incrociata: uno dei reni si sposta oltre la linea mediana. 7. Anomalie relazionali(fusione dei reni): rene a ferro di cavallo - fusione dagli stessi poli; rene a biscotto - fusione con bordi mediali; Rene a forma di S: il polo superiore di un rene è fuso con il polo inferiore dell'altro. 8. Policistico-molte piccole cisti nelle piramidi renali. A volte si combina con cisti nel fegato, nella milza, nel pancreas, nelle gonadi, nelle ossa ed è accompagnata da malformazioni agli occhi. 9. Idronefrosi congenita- la presenza di ostacoli al deflusso dell'urina (rene a ferro di cavallo, distopia) crea le condizioni per la ritenzione di urina nei tubuli urinari e nell'albero escretore. L'idronefrosi è caratterizzata dalla dilatazione del sistema pielocaliceale, dall'atrofia del parenchima renale e dal progressivo deterioramento della capacità funzionale dei reni a causa del ridotto deflusso delle urine.
Anomalie ureterali: 1. Diverticolo. 2. Valvole congenite ureteri. 3. Raddoppio ureteri. 4. Osti ectopici uretere - apertura dell'uretere negli organi cavi (vescicole seminali, utero, vagina).
Anomalie della vescica: 1. Ectopia vescica - manca la parete anteriore della vescica; sulla parete addominale anteriore è presente una fessura attraverso la quale sporgono la parete posteriore della vescica con un triangolo ed entrambe le aperture degli ureteri. 2. Fistola uracale- mancata chiusura del dotto urinario germinale. In questo caso, l'urina viene rilasciata dall'ombelico. 3. Doppia vescica. 4. Diverticoli Vescia.
Anomalie dell'uretra: 1. Atresia. 2.Raddoppio. 3. Ipospadia- una fessura dell'uretra maschile sulla sua parete ventrale (posteriore). 4. Epispadia- uno spazio congenito sulla parete dorsale (anteriore) dell'uretra maschile.
Genitali
Genitali maschili si dividono in interni (testicolo, epididimo, vasi deferenti, vescicole seminali, prostata) ed esterni (pene e scroto).
Testicolo(lat. testicolo, greco orchidea, didimo) - una ghiandola maschile accoppiata - svolge due importanti funzioni: produce sperma ("secrezione esterna") e ormoni sessuali ("secrezione interna"), che influenzano lo sviluppo dei caratteri sessuali primari e secondari. Il testicolo si trova nello scroto e ha due estremità ( estremità superiore e inferiore), due superfici ( facies laterale e mediale) e due bordi ( margo anteriore e posteriore). Il testicolo sinistro è solitamente abbassato leggermente più in basso di quello destro. Ciò è dovuto al fatto che la vena testicolare sinistra scorre nella vena renale ad angolo retto e la vena testicolare destra scorre nella vena cava inferiore ad angolo acuto, cioè il deflusso venoso a sinistra è alquanto difficile. Questa caratteristica anatomica del deflusso venoso spiega anche il fatto che i varicoceli (vene varicose del funicolo spermatico) si osservano più spesso a sinistra che a destra. L'esterno del testicolo è ricoperto da una fitta tunica albuginea ( tunica albuginea), che penetra in esso lungo il bordo posteriore e forma il mediastino (mediastino). Da quest'ultimo si estendono a ventaglio i setti che dividono il testicolo in lobuli. Ciascun lobulo contiene da 1 a 3 tubuli seminiferi contorti ( tubuli seminiferi contorti), che contengono epitelio spermatogenico. I tubuli contorti di un lobulo si fondono tra loro e formano un tubulo seminifero dritto (ce ne sono da 100 a 300 nel testicolo), che entrano nel mediastino del testicolo e formano una rete ( rete testicolare). Da questa rete emergono 12-15 tubuli efferenti che formano un dotto epididimale molto fortemente contorto, lungo 6-8 m, nel quale avviene solo la maturazione finale degli spermatozoi.
Inizia dalla coda dell'appendice dotto deferente (Dotto deferente), che, come parte del cordone spermatico, passa attraverso il canale inguinale, e poi lungo la parete laterale del bacino verso il basso e indietro, dirigendosi verso il fondo della vescica. La sezione terminale del condotto si espande formando un'ampolla a forma di fuso. Sulla parete posteriore della vescica sono presenti vescicole seminali, la cui secrezione, mescolandosi con lo sperma, le liquefa, le nutre e le attiva. Condotto delle vescicole seminali ( dotto escretorio) si collega ai dotti deferenti sotto l'ampolla e si forma dotto deferente (dotto eiaculatorio), che si apre nella parte prostatica dell'uretra sul collicolo seminifero su entrambi i lati dell'utero prostatico.
Cordone spermatico (funicolo spermatico) - un complesso di formazioni risultanti dalla discesa del testicolo dalla cavità addominale nello scroto. Il cordone spermatico inizia dall'estremità superiore del testicolo, passa nel canale inguinale e termina nella regione dell'anello inguinale profondo. Il cordone spermatico comprende una serie di strutture (fascia, vene e plessi venosi, arterie, muscoli, nervi e vasi linfatici) Prostata (prostata) - un organo ghiandolare-muscolare spaiato, figurativamente "il secondo cuore di un uomo". La sua funzione come ghiandola è quella di secernere una secrezione che nutre e attiva lo sperma. La contrazione del tessuto muscolare della ghiandola impedisce all'urina di entrare nell'uretra durante l'eiaculazione. Le ghiandole formano il parenchima ghiandolare ( parenchima ghiandolare), il tessuto muscolare costituisce la sostanza muscolare ( substantio muscolare). L'esterno della ghiandola è ricoperto da una capsula di tessuto connettivo. La ghiandola prostatica si trova sotto la vescica sul diaframma urogenitale. La sua forma e dimensione ricordano una castagna. La parte prostatica dell'uretra passa attraverso la ghiandola. È costituito da tre lobi: destro, sinistro e medio. La parte della ghiandola prostatica compresa tra entrambi i dotti eiaculatori (ductus ejaculatorius) e la superficie posteriore dell'uretra costituisce il lobo medio o istmo ( istmo).
Pene(lat. pene- coda, greco fallo impudico- spugnola vergognosa) serve per espellere l'urina ed espellere lo sperma, essendo un organo di copulazione. Il dorso del pene è formato da due fusi cavernoso (cavernoso) corpi che iniziano davanti con estremità appuntite e dietro divergono in gambe attaccate ai rami inferiori delle ossa pubiche. Sul lato della superficie uretrale ce n'è un terzo cavernoso (spugnoso) il corpo, che all'estremità distale del pene passa in una formazione a forma di cono - Testa pene. La base della testa è approfondita e, come un cappuccio, copre la parte anteriore a forma di cono dei corpi cavernosi (cavernosi) collegati. I bordi del glande e della sua corolla sporgono oltre l'estremità anteriore dei corpi cavernosi verso la radice dell'organo. La pelle che ricopre il corpo dell'organo è sottile, facilmente rimovibile, pigmentata sulla superficie uretrale e priva di grasso sottocutaneo. La pelle del corpo entra in una piega libera prepuzio che copre la testa del pene. Briglia- una piccola piega mediana sulla superficie dell'uretra - collega il prepuzio con la pelle del glande, che in apparenza ricorda più la mucosa. Alla radice del pene si separano i suoi tre corpi costituenti. I corpi cavernosi divergono in direzioni diverse e ciascuno di essi, smussandosi gradualmente, viene infine attaccato mediante tessuto fibroso al periostio delle ossa pubiche. Ogni gamba è coperta muscolo ischiocavernoso. Il corpo spugnoso si trova sotto e tra i corpi cavernosi e si ispessisce all'estremità formando un ispessimento sferico chiamato bulbo del pene. Questa istruzione è coperta muscolo bulbocavernoso. Nessuno di questi muscoli striati è coinvolto nel processo di erezione, ma tutti si contraggono ritmicamente durante l’eiaculazione per aiutare a spingere lo sperma. Tutti e tre questi cilindri sono racchiusi in una guaina fasciale, costituita da due fasce: superficiale e profonda. Inoltre, i corpi cavernosi (cavernosi) - le principali strutture che facilitano l'erezione - sono racchiusi in una capsula fibrosa spessa e densa - tunica albuginea, che nella sua sezione centrale forma il setto del pene. Va sottolineato che il setto lungo gran parte della lunghezza del pene non separa completamente i corpi cavernosi, ma prima che divergano sotto forma di zampe, ne garantisce la completa separazione. Fili di tessuto fibroso si estendono dalla superficie interna della tunica albuginea ( trabecole). Il corpo spugnoso del pene è racchiuso nella propria tunica albuginea, più sottile della tunica albuginea dei corpi cavernosi. Anche il tessuto del bulbo e la parte uretrale del corpo dell'organo rappresentano una struttura cavernosa, ma gli spazi interni sono più ordinati e le trabecole contengono più fibre elastiche che nei corpi cavernosi. Questa struttura è ideale per garantire il libero passaggio dei liquidi nell'uretra. L'afflusso di sangue al pene è fornito dalle arterie uretrale, bulbare, profonda e dorsale (rami dell'arteria pudenda interna), nonché dall'arteria pudenda esterna. Il deflusso venoso avviene attraverso la vena dorsale profonda (scorre nel plesso venoso prostatico) e la vena dorsale superficiale (scorre nella v. safena magna).
Scroto (scroto) è come un “termostato fisiologico” che mantiene la temperatura dei testicoli ad un livello inferiore a quella corporea. Questa è una condizione necessaria per la normale spermatogenesi. Si forma a seguito della discesa testicolare dovuta a strati parete addominale anteriore: 1) pelle; 2) dal grasso sottocutaneo, privo di grasso - la membrana carnosa (tunica dartos); 3) dalla fascia superficiale - fascia spermatica esterna (fascia spermatica esterna); 4) dalla fascia intercruralis - fascia del muscolo che solleva il testicolo (fascia cremasterica); 5) dal muscolo obliquo interno dell'addome e trasverso (m.obliqus internus abdominis et m.transversus abdominis) - il muscolo che solleva il testicolo (m.cremaster); 6) dalla facies trasversale (fascia transversalis) - fascia spermatica interna (fascia spermatica interna); 7) dal processo vaginale (processus vaginalis) del peritoneo - la membrana sierosa del testicolo (tunica vaginalis testis).
Organi genitali femminili si dividono in interne (ovaio, tube di Falloppio, utero, vagina) ed esterne (grandi e piccole labbra, vestibolo della vagina e clitoride).
Ovaio (ovario, ooforo) - un organo accoppiato dove avviene la formazione e la maturazione della cellula riproduttiva femminile (ovulo). L'ovaio è una ghiandola endocrina in cui vengono prodotti gli ormoni sessuali femminili. L'ovaia si trova sulla parete laterale del bacino. Ha la forma di un'ellisse appiattita. La sua lunghezza è di 2,5 - 5 cm; larghezza - 1,5 - 3 cm; spessore 0,5 - 1,5 cm Con la cessazione delle mestruazioni nelle donne di età compresa tra 40 e 50 anni, si verifica l'atrofia ovarica e le loro dimensioni diminuiscono di circa la metà. Vicino all'ovaio si trovano le sue appendici, che sono formazioni rudimentali (resti di dotti mesonefrici): appendice ovaia (epoforone) E periovarico (paraoforone), costituito da numerosi tubuli ciechi, si trova nel mesentere della tuba di Falloppio. L'ovaio ha una superficie mediale rivolta verso la cavità pelvica e una superficie laterale adiacente alla parete pelvica. Si distinguono il bordo libero e il bordo mesenterico, su cui si trova la porta dell'ovaio e è attaccato il mesentere, un duplicato del peritoneo, proveniente dallo strato posteriore del legamento largo dell'utero. Dall'estremità uterina dell'ovaio ( estremità uterina) va all'utero con il proprio legamento dell'ovaio, situato tra le due foglie del legamento largo dell'utero, e l'estremità tubarica ( extremitas tubaria) rivolto verso le tube di Falloppio. Nonostante l'ovaio abbia un mesentere, non è ricoperto di peritoneo, ma di un epitelio germinale a strato singolo, sotto il quale si trova il tessuto connettivo tunica albuginea ( tunica albuginea). All'interno di esso c'è una corteccia costituita da tessuto connettivo, in cui sono presenti numerosi follicoli: primari, in crescita (in maturazione), atresici (in fase di sviluppo inverso), nonché corpo luteo e cicatrici. Il midollo dell'ovaio è formato da tessuto connettivo in cui passano vasi sanguigni e nervi. A differenza delle cellule germinali maschili, le cellule riproduttive femminili si riproducono nel periodo prenatale, determinando la formazione di follicoli primari contenenti un ovulo. Una neonata ha fino a 800mila follicoli primari in entrambe le ovaie. Il loro numero non solo non aumenta dopo la nascita, ma diminuisce rapidamente a causa del riassorbimento. Al momento della pubertà, nella corteccia rimangono solo 400-500 follicoli primari, che si trasformano in follicoli maturi - vescicolari ( Bolle di Graaf). Dopo l'ovulazione - il rilascio dell'ovulo nella cavità addominale libera - si sviluppa al posto della vescicola di Graaf corpo luteo(mestruale o di gravidanza) producendo ormone progesterone, che è responsabile della preparazione dell'epitelio uterino per l'impianto dell'uovo e le prime fasi dell'allattamento.
Ovidotto(Fallopieva) ( tuba uterina, salpinge, oovidotto) - un organo accoppiato che serve a trasportare l'uovo dall'ovaio nella cavità uterina ed è il sito di fecondazione dell'uovo. Le tube di Falloppio si trovano nella cavità pelvica su entrambi i lati del fondo uterino, nel bordo superiore del legamento largo dell'utero. La parte del legamento largo dell'utero tra le tube di Falloppio e l'ovaio è chiamata mesentere delle tube di Falloppio ( mesosalpinge). Le tube di Falloppio contengono quanto segue: parti: 1. Parte uterina- la parte uterina, situata nella parete dell'utero; 2. Istmo tubae uterinae- l'istmo delle tube di Falloppio è la parte più spessa e stretta; 3. Ampolla tuba uterina- ampolla della tuba di Falloppio, che è adiacente alla superficie mediale dell'ovaio; 4 . Infundibulum tubae uterinae- l'imbuto delle tube di Falloppio, che termina con le fimbrie delle tube, una delle quali è più lunga delle altre, raggiunge l'ovaio e vi si accresce. Secondo la fimbria ovarica, l'uovo rilasciato dall'ovaio entra nel lume della tuba di Falloppio. La parete delle tube di Falloppio è costituita da tre conchiglie: 1. Tunica sierosa- membrana sierosa, che è la parte superiore del legamento largo dell'utero; 2. Tunica muscolare- lo strato muscolare è rappresentato da due strati (longitudinale e circolare), che sono una continuazione dei muscoli dell'utero. Lo strato muscolare nell'imbuto della tuba di Falloppio si assottiglia, più vicino all'utero si ispessisce; 3. Mucosa della tonaca- la mucosa è costituita da un epitelio ciliato monostrato, le cui ciglia sfarfallano verso l'estremità uterina del tubo, favorendo così l'avanzamento dell'uovo nella cavità uterina.
Utero(lat. utero, greco metra, isteria) - un organo muscolare cavo spaiato svolge la funzione mestruale, in cui si sviluppa l'embrione e nasce il feto. L'utero si trova nella pelvi, il suo fondo espanso ( fondo) rivolto verso l'alto, seguito da un corpo ispessito ( corpo) e il collo ristretto ( cervice) si apre nella vagina con un'apertura limitata dalle labbra anteriori e posteriori. Pertanto, nella cervice si distingue la parte sopravaginale ( porticato sopravaginale) e la parte vaginale ( porticato vaginale). Inoltre, la cervice è coperta archi anteriori e posteriori vagina. Inoltre, quello posteriore è più profondo ed è adiacente al punto più basso della cavità peritoneale: la sacca di Douglas. (excavatio rectouterina). Questa caratteristica strutturale anatomica è utilizzata nella medicina pratica. Vale a dire, quando si verifica un processo patologico di eziologia sconosciuta nella cavità peritoneale, viene eseguita una colpotomia diagnostica (dissezione del fornice posteriore), poiché l'essudato (sangue, pus, ecc.) Si accumulerà nella sacca di Douglas. La superficie anteriore dell'utero è rivolta verso la vescica, la superficie posteriore è rivolta verso il retto.
Posizione La dimensione dell'utero nella cavità pelvica dipende dal grado di riempimento degli organi vicini e dall'apparato di fissazione. Con un leggero riempimento della vescica e del retto, l'utero di una donna non incinta si trova in modo tale che le bocche delle tube di Falloppio siano simmetriche rispetto al piano mediosagittale e l'utero stesso sia inclinato in avanti ( antiverso). Inoltre, tra il corpo e il collo si forma un angolo aperto anteriormente: il corpo è piegato anteriormente rispetto al collo ( anteFlessico), quindi il fondo dell'utero giace sulla vescica. Quando la vescica è piena, l'angolo tra la cervice e il corpo dell'utero viene levigato, il fondo dell'utero si solleva. In alcuni casi, l’utero è inclinato all’indietro ( retroversione) e può anche essere curvato posteriormente ( retroflessio). Questa posizione dell'utero è considerata patologica.
Molto importante nella sintopia degli organi pelvici è il rapporto degli ureteri con l'utero e l'arteria uterina. Gli ureteri penetrano nella pelvi, diffondendosi attraverso i vasi iliaci, con l'uretere sinistro che attraversa l'aliaca communis e quello destro che attraversa l'iliaca esterna. Sotto, gli ureteri sono incrociati dall'interno n. e vasa obturatoria e a livello della metà dell'utero ad una distanza di 1-2 cm da esso si intersecano con a.uterina. Va ricordato che l'arteria passa davanti all'uretere. Questo crossover è molto importante durante le operazioni di rimozione dell'utero, perché a volte l'uretere rimane intrappolato in una pinza insieme all'arteria uterina, che in questo caso può essere tagliata accidentalmente.
La parete dell'utero è costituita da tre conchiglie: 1. Membrana sierosa ( perimetro) è rappresentato dal peritoneo, che ricopre l'utero su tutti i lati tranne la parte vaginale della cervice. Gli strati del peritoneo passano nei legamenti larghi destro e sinistro dell'utero, che continuano nello strato parietale del peritoneo pelvico. Tra le foglie si trova il legamento largo dell'utero legamento rotondo dell'utero, che ha origine dalla superficie laterale dell'utero appena sotto l'imboccatura delle tube di Falloppio, segue lateralmente verso il basso, passa attraverso il canale inguinale e raggiunge il pube, dove le sue fibre si intrecciano nel tessuto. Nella zona delle superfici laterali della cervice tra i due strati del legamento largo è presente tessuto periuterino ( parametrio), in cui passano l'uretere, l'arteria uterina, la vena e le fibre nervose. Ecco perché la puntura del fornice laterale e altre manipolazioni chirurgiche in quest'area devono essere eseguite in modo estremamente estremo accuratamente. 2. Membrana muscolare ( miometrio) è formato da tessuto muscolare non striato (liscio), rappresentato da strati circolari longitudinali e medi interni ed esterni, intrecciati tra loro. Durante la gravidanza, le fibre muscolari si ipertrofizzano, la loro dimensione aumenta di 5-10 volte in lunghezza e 3-4 volte in larghezza, e la dimensione dell'utero aumenta di conseguenza. 3. Mucosa ( endometrio) è ricoperto da un epitelio colonnare monostrato; nella lamina propria ben definita sono presenti numerose ghiandole uterine tubolari semplici. La mucosa del canale cervicale è composta prevalentemente da cellule cilindriche alte che producono muco, da cui si forma un tappo mucoso che impedisce all'infezione di entrare nell'utero, nelle tube di Falloppio e nella cavità peritoneale.
I legamenti dell'utero, che corrono in tutte le direzioni, fissando l'utero all'osso sacro, alla sinfisi pubica e alle pareti laterali della parete pelvica, sono più pronunciati nella cervice. L'attività fisica intensa, il parto frequente e una serie di altri motivi sono una conseguenza dell'indebolimento dell'apparato di fissaggio, che porta al prolasso uterino.
Vagina (vagina, colpi) è un tubo spaiato lungo 7-10 cm, appiattito dalla parte anteriore a quella posteriore, che comunica con l'utero in alto, passa attraverso il diaframma urogenitale in basso e si apre nel vestibolo della vagina con un'apertura ( ostio vaginale), dove è ricoperto dall'imene ( imene) o i suoi resti. Si distingue la parete anteriore della vagina, che nel terzo superiore è adiacente alla vescica, e nella restante area è fusa con la parete dell'uretra femminile. La parete posteriore nella sua parte superiore è ricoperta di peritoneo e nella parte inferiore è adiacente alla parete anteriore del retto. A causa del fatto che i setti descritti sono piuttosto sottili, in caso di lesioni traumatiche o durante il parto, oppure in seguito a processi infiammatori, si possono formare fistole, che spesso non guariscono per lungo tempo, tra la vagina e l'adiacente organi.
Parete vaginaleè costituito dalle membrane mucose, muscolari e avventiziali. Le cellule dello strato superficiale dell'epitelio della mucosa sono ricche di glicogeno che, sotto l'influenza dei microbi che vivono nella vagina, si decompone per formare acido lattico. Ciò conferisce al muco vaginale una reazione acida e lo rende battericida contro i microbi patogeni. La copertura muscolare è rappresentata prevalentemente da fasci di fibre muscolari orientati longitudinalmente, nonché da fasci che hanno una direzione circolare. Nella parte superiore, lo strato muscolare passa nei muscoli dell'utero, nella parte inferiore diventa più potente ed entra in contatto con i muscoli del perineo. Fasci di muscoli striati del diaframma urogenitale, che coprono l'estremità inferiore della vagina e allo stesso tempo l'uretra, formano una sorta di sfintere muscolare. Il pavimento pelvico, costituito da un massiccio tessuto muscolo-fibroso, è della massima importanza nel fissare la vagina. Quando il pavimento pelvico è indebolito nelle donne anziane, sotto l'influenza di un aumento prolungato della pressione intra-addominale, si può osservare a vari livelli prolasso della parete vaginale, prolasso vaginale accompagnato da prolasso o addirittura prolasso dell'utero.
Grandi labbra (grandi labbra pudendi) sono pieghe cutanee a forma di rullo delimitate lateralmente dalla pelle della coscia dal solco femoro-perineale. Davanti e dietro, entrambe le grandi labbra sono collegate da aderenze ( commissurae labiorum posteriore e anteriore). Genitali minori labbra (piccole labbra pudendi) sono costituiti da tessuto connettivo, situato verso l'interno delle grandi labbra nella fessura genitale, che delimita il vestibolo della vagina. I bordi anteriori delle piccole labbra sono liberi. Quelli posteriori sono collegati tra loro e formano una briglia ( frenulo labiorum pudendi). L'estremità superiore di ciascuna piccola labbra si divide in due gambi che conducono verso il clitoride. La gamba laterale circonda lateralmente il clitoride e lo copre dall'alto, formando il prepuzio del clitoride ( prepuzio clitoride). La cru mediale si avvicina al clitoride dal basso e si fonde con la cru del lato opposto, formando il frenulo del clitoride (frenulum clitoridis). Il vestibolo della vagina è limitato lateralmente dalle superfici mediali delle piccole labbra, anteriormente dal clitoride e posteriormente dalla fossa del vestibolo vaginale. Alla base delle piccole labbra, i dotti delle grandi ghiandole vestibolari si aprono nel vestibolo della vagina ( ghiandole vestibolari maggiori), che secernono un liquido simile al muco che idrata la parete dell'ingresso della vagina e i dotti delle piccole ghiandole vestibolari - nella parete del vestibolo. Inoltre, la vagina e l'apertura esterna dell'uretra, che si trova tra il clitoride e l'ingresso della vagina, si aprono nel vestibolo della vagina. Il bulbo del vestibolo si proietta tra l'apertura dell'uretra e il clitoride, le cui parti laterali si trovano alla base delle grandi labbra, adiacenti alle grandi ghiandole del vestibolo. Il bulbo del vestibolo è identico al corpo spugnoso spaiato del pene. Clitorideè identico ai corpi cavernosi (cavernosi) del pene.
Le anomalie dello sviluppo del sistema genito-urinario sono un gruppo di malattie congenite, la cui particolarità è una malformazione intrauterina del feto, a seguito della quale sono colpiti uno o più organi del sistema genito-urinario. Malattie di questo tipo sono molto comuni e rappresentano circa il 40% di tutti i disturbi dello sviluppo che si verificano in utero.
Patologie della vescica
Tra le anomalie congenite, l'estrofia di questo organo è considerata molto grave e pericolosa. Questa malattia, come altre anomalie nello sviluppo della vescica, è spesso accompagnata da altre anomalie dell'intero sistema (tra cui patologie della formazione dei reni, delle ossa pubiche o dell'uretra). Non per niente questa malattia è considerata un tipo estremamente grave di anomalia della formazione del feto all'interno dell'utero, perché a causa del suo sviluppo, la vescica non ha una parete anteriore e non esiste nemmeno una parete addominale anteriore, che provoca perdite continue di urina, nonché la comparsa di ulcere sulla superficie del perineo e dei genitali. Questa malattia colpisce 1 su 30-40mila neonati. Le donne hanno il doppio delle probabilità di sviluppare questa patologia. Inoltre, se il primo figlio è nato con una tale malattia, in 1 caso su 100 anche il secondo soffrirà di una malattia simile.
Una malattia congenita molto comune associata all'organo in questione è il diverticolo della vescica. In questo caso, una delle pareti sporgerà verso l'esterno. Il diverticolo può essere non solo congenito, ma anche acquisito in età adulta. Inoltre, ci sono diverticoli singoli e multipli (differenti, rispettivamente, nel numero di sporgenze), nonché falsi e veri (la sporgenza ha tutti gli strati caratteristici della parete di questo organo secondo la sua istologia e con un falso uno è presente una protuberanza erniaria della mucosa che urta il tessuto muscolo-vescicale).
È comune anche l’agenesia vescicale. Quando si sviluppa questa malattia, l’organo non è affatto presente nel corpo del neonato. In un numero significativo di casi, tali deviazioni dello sviluppo sono accompagnate da altre anomalie, che insieme portano alla morte a causa di problemi con le funzioni vitali del corpo.
L'esatto opposto dell'agenesia della vescica è il suo raddoppio. In tali circostanze, piuttosto rare, l'organo si sviluppa con un setto al suo interno, dividendolo in due componenti separate. In entrambe queste cavità si apre l'orifizio indipendente dell'uretere. In alcuni casi, questo difetto è accompagnato dalla presenza di uretra e cervici in duplice copia. È interessante notare che in rari casi il setto non si sviluppa completamente e serve a formare la cosiddetta “vescica a due camere”.

Ma anche le malattie di questa categoria comprendono la contrattura del collo della vescica. Quando si sviluppa questo scenario, si forma troppo tessuto fibroso nella parete della vescica nella zona cervicale. A seconda della quantità di tessuto in eccesso prodotto, la fibrosi derivante dalla contrattura cervicale può avere diversi gradi di gravità. Nei casi lievi, i pazienti riscontrano solo lievi problemi durante la minzione. Quando la fibrosi è in una fase grave, il processo di fuoriuscita di liquidi dalla vescica viene interrotto. La manifestazione di tale deviazione può essere il reflusso vescico-ureterale e la progressione dell'infiammazione (ad esempio uretrite) nel sistema urinario a causa della penetrazione dell'infezione urogenitale.
Inoltre, esistono diversi tipi di patologia uracale. Questo organo è presente nel nostro corpo durante lo sviluppo fetale e serve a collegare la vescica e il liquido amniotico. In una situazione normale, al momento della nascita di una persona, l'uraco è completamente retratto. Altrimenti si può sviluppare una delle patologie legate al rilascio di urina attraverso l'apertura del cordone ombelicale.
Anomalie degli organi genito-urinari
Le più comuni sono le anomalie dello sviluppo degli ureteri. Le malattie di questa categoria costituiscono circa un quinto di tutti i disturbi legati ai disturbi del sistema genito-urinario. Come altre anomalie nel campo dell'urologia, questi disturbi dello sviluppo in molti casi vengono rilevati accidentalmente nei bambini durante esami relativi ad altre malattie e talvolta sono anche accompagnati da disfunzione del sistema renale. Ai pazienti viene spesso diagnosticato un restringimento o una dilatazione dell'uretere. Nonostante l'enorme numero di diversi tipi di anomalie associate all'uretere, queste causano disturbi nell'urodinamica (il movimento dell'urina attraverso il sistema urinario). Di conseguenza, l'ostruzione del flusso di urina porta allo sviluppo di processi infiammatori nei reni (ad esempio la cistite), che causa l'interruzione del suo funzionamento. In molti casi, ai bambini vengono diagnosticate diverse anomalie degli ureteri su entrambi i lati, che portano a cambiamenti irreversibili nel rene. Tipicamente, tali malformazioni vengono rilevate tra i 6 ei 10 anni.

Le anomalie nello sviluppo degli organi genitali maschili si riferiscono a difetti nella formazione delle parti genito-urinarie del corpo. Molto spesso, in questa categoria è incluso lo sviluppo anormale del testicolo, con il quale nasce dal 5 al 7% dei bambini. Tra tali deviazioni si distinguono diverse quantitative: anarchismo (in cui entrambi i testicoli sono completamente assenti), monorchismo (ce n'è solo uno), poliorchismo (il numero è tre o più). Tra le altre anomalie dello sviluppo testicolare si notano ipoplasia (sottosviluppo) di questo organo e criptorchidismo (non scende nello scroto). L'ultima anomalia è considerata la più frequente di tutte. Le malattie di questa categoria sono monitorate con molta attenzione dai medici, poiché le loro conseguenze determinano se un ragazzo potrà avere figli da adulto.

Agenesia renale
Le anomalie dello sviluppo renale sono un altro tipo di malformazione degli organi genito-urinari. I problemi con la loro formazione sono tipici di un terzo di tutti i pazienti con malattie urologiche. A causa del frequente rilevamento di tali anomalie e delle gravi conseguenze di malattie renali associate a difetti dello sviluppo, i medici sono sempre più attenti ai segnali che possono indicare la presenza di eventuali problemi. In molti casi, un trattamento tempestivo aiuta a prevenire lo sviluppo di malattie molto più gravi ed evitare conseguenze gravi e talvolta irreversibili. In molti casi, i disturbi dello sviluppo renale sono accompagnati da anomalie nello sviluppo dei vasi renali. A causa del fatto che i reni con difetti dello sviluppo hanno disturbi della circolazione sanguigna e dell'urodinamica, nel corpo iniziano a formarsi processi infiammatori cronici. Ciò porta al fatto che i reni con patologie strutturali sono più vulnerabili a malattie come pielonefrite, idronefrosi e calcoli renali. È importante sottolineare che in assenza di sintomi e altre manifestazioni evidenti della malattia, a volte le anomalie non vengono rilevate per tutta la vita.
Oltre alle anomalie nello sviluppo dei reni, i medici distinguono tra problemi relativi alla formazione dei vasi renali. Patologie di questo tipo sono comuni. La gravità di tale disturbo dipende dal fatto che sia accompagnato da una malattia renale o se sia l'unico tipo di anomalia del sistema genito-urinario. In alcuni casi, vasi renali anormali portano a vari disturbi renali, tuttavia, la maggior parte di tali disturbi non si manifesta.
Cause di anomalie
Molto spesso, le malattie del sistema genito-urinario associate ad anomalie dello sviluppo sono causate da una serie di fattori accertati. Questi includono:
- predisposizione genetica (la malattia viene trasmessa a livello genetico);
- malattie infettive e veneree subite durante la gravidanza;
- esposizione alle radiazioni, nonché alle radiazioni ionizzanti;
- dipendenza da alcol e prodotti del tabacco;
- dieta sbagliata;
- contatto con sostanze tossiche;
- uso di farmaci ormonali.
Se viene rilevata un'anomalia nello sviluppo del sistema genito-urinario, è richiesto un esame di tutti i membri della famiglia del paziente. Inoltre, la consultazione con uno specialista aiuterà a chiarire la situazione e a calcolare i rischi di ripetere uno scenario simile nelle generazioni future.

Una delle anomalie di maturazione più comuni all'interno dell'utero, caratteristica delle vie urinarie superiori, è la loro duplicazione. Per lo più è localizzato su un lato, in casi più rari è presente su entrambi. In una situazione con duplicazione del tratto urinario superiore, spesso ci sono altri due difetti degli orifizi ureterali: questi includono l'ectopia dell'orifizio ureterale e l'ureterocele. In generale, le patologie delle vie urinarie superiori si dividono in quattro gruppi abbastanza ampi:
- anomalie associate ad alterata comunicazione dei vasi sanguigni con la parte superiore delle vie urinarie;
- displasia parziale della parete ureterale;
- ectopia degli orifizi ureterali;
- ureterocele.
Inoltre, tra le malformazioni congenite delle vie urinarie rientrano tutte le malattie associate ad anomalie nello sviluppo dei reni. Un tale disturbo, ad esempio, è il raddoppio dei reni. Questa anomalia spesso diagnosticata è caratterizzata dalla duplicazione di alcuni componenti del rene (bacino, vasi o ureteri) o da tutti questi elementi contemporaneamente, fenomeno chiamato duplicazione completa del rene. In questo caso, entrambe le metà sono generalmente considerate organi indipendenti e il processo patologico spesso colpisce solo uno degli organi.
Le seguenti anomalie dello sviluppo sono caratteristiche dei reni: malposizione e distopia. Questi sottotipi di anomalie si dividono in pelviche, iliache, lombari, toraciche e trasversali. Allo stesso tempo, i pazienti affetti da distopia hanno spesso problemi con l'attività intestinale. In generale, senza determinate manifestazioni cliniche, questi tipi di malattie possono rimanere inosservate per un periodo di tempo molto lungo.
Una delle gravi deviazioni nella formazione delle vie urinarie è considerata un'anomalia nella relazione tra i reni, in cui gli organi crescono insieme. A seconda del tipo di fusione possono assumere forme diverse, modificando l'aspetto dell'organo a forma di L, a S e a ferro di cavallo. I reni fusi tra loro sono facilmente soggetti a processi infiammatori e inoltre possono causare ipertensione renale. A causa dello sviluppo di altre malattie causate da processi infiammatori (pielonefrite, idronefrosi, afflusso sanguigno anomalo), anche l'ipertensione arteriosa può progredire.

Un’altra anomalia dello sviluppo grave è la malattia del rene policistico. La malattia viene spesso trasmessa geneticamente e ha un tipo di ereditarietà dominante. Con questo disturbo, la maggior parte del parenchima renale è affetto da un numero enorme di tumori cavi (cisti) di diverse dimensioni. In 7 casi su 10, un bambino i cui organi sono affetti dalla malattia policistica nasce morto. Se il numero di tumori non è così elevato, la loro prestazione (e quindi la vitalità dell'organismo) viene mantenuta, ma in molti casi esiste un'alta probabilità di sviluppare insufficienza renale. Questa malattia è spesso accompagnata dalla malattia policistica di altre parti del corpo.
Un'altra anomalia non ereditaria è una cisti solitaria dell'organo in questione. Questa malattia coinvolge una singola cisti nel rene, che può essere congenita o acquisita. Con l’aumento delle dimensioni della cisti, la funzione renale viene compromessa e i pazienti soffrono di dolore lombare.
L'anomalia nota come rene spugnoso è caratterizzata dalla formazione di un gran numero di tumori nelle piramidi renali. Questa malattia di solito non porta allo sviluppo di insufficienza d'organo.
Separatamente, vale la pena considerare l'ureterocele: questa è una sporgenza, più simile a un'ernia, all'interno dell'uretere. Dopotutto, a volte provoca la dilatazione delle vie urinarie superiori, la pielonefrite e l'urolitiasi.
Decorso della malattia

Malattia policistica. Cisti renale
Il decorso varia per ogni singola anomalia del sistema genito-urinario. Alcune di queste anomalie possono non manifestarsi per molto tempo e vengono scoperte in modo del tutto casuale durante esami relativi ad altri disturbi. Altri diventano prerequisiti per lo sviluppo di urolitiasi, insufficienza renale cronica e ipertensione arteriosa. In casi specifici, le patologie nella formazione degli organi del sistema genito-urinario diventano la causa della morte di un bambino nel grembo materno o in tenera età, che è associata a una violazione delle funzioni vitali del corpo. A volte tali anomalie sono caratterizzate da una progressione lenta, che alla fine influenza la manifestazione dei sintomi della malattia in età avanzata.
Diagnostica
Quando identificano le malformazioni del sistema genito-urinario, i medici utilizzano metodi diagnostici differenziati, che includono:
- radiografia con introduzione di liquido di contrasto;
- Risonanza magnetica;
- TAC;
- uretrografia retrograda;
- inserimento di un catetere nell'uretere;
- cistoscopia;
- ecografia.
La diagnosi tempestiva di malattie di questo tipo, prescritta in tenera età, è semplicemente necessaria. Ad esempio, nel caso di un testicolo ritenuto, l'esame ecografico aiuta a stabilirne con certezza la posizione, e l'assunzione di un liquido di contrasto durante l'esecuzione di una radiografia permette di determinare la presenza di aplasia renale o diverticolo vescicale.
Grazie alla diagnosi tempestiva e al trattamento necessario, è possibile evitare problemi, sia fisici che psicologici. Ad esempio, quando il pene è piegato, l'anomalia diventa causa di problemi con la minzione (più spesso riscontrati nei bambini), nonché una barriera psicologica nella sfera intima (tipica degli uomini adulti). Inoltre, questa malattia provoca infertilità (che deriva anche dal criptorchidismo bilaterale). Ad esempio, il criptorchidismo, la cui diagnosi e trattamento sono stati ritardati, provoca la torsione testicolare.
Trattamento delle anomalie del sistema genito-urinario
Spesso le patologie richiedono un intervento chirurgico nel corpo. Tali operazioni variano in volume e complessità. In caso di criptorchidismo (testicolo ritenuto nello scroto), l'intervento chirurgico necessario può essere eseguito sia nell'infanzia che nell'età adulta. Alcuni interventi medici sono prescritti esclusivamente nei casi in cui i metodi tradizionali non portano alcun risultato.
Le più difficili da trattare e correggere sono le anomalie legate all'ermafroditismo. In questi casi, si consiglia ai pazienti di ricorrere alla chirurgia. La terapia ormonale che corrisponde alla definizione di genere di una persona non sarà superflua.
Il tratto urinario inizia con la pelvi renale e termina con l'apertura uretrale.
Malformazioni congenite (anomalie) delle vie urinarie
Le anomalie congenite del tratto urinario sono strettamente correlate alle anomalie renali identiche. Se un paziente ha due reni, ci sono tre ureteri su un lato. In caso di agenesia renale unilaterale, l'uretere stesso è corrispondentemente assente. Insieme alla presenza di un doppio rene, è abbastanza comune anche l'uretrocele: sporgenza dell'uretra attraverso la parete vaginale, con conseguente formazione di un gonfiore bulboso. L'uretere può presentare dei restringimenti, eventualmente anche nel caso di un ramo cieco. Inoltre, è possibile che l'uretere non finisca nell'uretere, ma, ad esempio, sfoci nelle vescicole seminali o nell'uretra stessa.
Malformazioni congenite (anomalie) della vescica
Quando il dotto urinario non viene chiuso durante lo sviluppo, molto spesso inizia a formarsi una cisti. È anche possibile formare un diverticolo della vescica, una sporgenza a forma di imbuto o simile a una sacca della parete dell'organo. Tutte le anomalie di cui sopra non sono accompagnate da malattia. Una delle malformazioni più gravi delle vie urinarie è l'estrofia della vescica, che si manifesta con danni non solo alla vescica, ma anche alla parete anteriore del peritoneo, dell'uretra e delle ossa pelviche. Se non esiste un trattamento per queste anomalie, il paziente muore.
Anomalie congenite dell'uretra
L'epispadia è un difetto dello sviluppo (anomalia) del pene, in cui l'apertura dell'uretra, destinata alla fuoriuscita dell'urina, si trova nella parte superiore del pene. Anche le donne presentano spesso malformazioni della vescica e del clitoride. L'ipospadia è una malattia in cui l'apertura esterna (escretoria) dell'uretra si trova nella parte inferiore del pene.
Sintomi di anomalie renali:
disturbo urinario;
disturbo della crescita;
dolore al fianco e all'addome;
infezioni frequenti delle vie urinarie.

Cause di anomalie del tratto urinario
Tutte le suddette anomalie del sistema di estrazione dell'urina dal corpo sono congenite. Di conseguenza, possono insorgere a causa di un difetto genetico o come risultato dell'esposizione a fattori dannosi sul feto durante la gestazione.
Trattamento delle anomalie del sistema genito-urinario
Nella maggior parte dei casi, il trattamento è chirurgico. Naturalmente, quando l'anomalia congenita non interferisce con il flusso dell'urina, il trattamento non è estremamente necessario.
L'automedicazione in presenza di anomalie congenite delle vie urinarie è severamente vietata.
La diagnosi di anomalie nei bambini piccoli è estremamente difficile. I genitori dovrebbero ricordare i principali sintomi delle anomalie congenite di questo sistema corporeo e monitorarli man mano che il bambino cresce. Questo è un aumento della temperatura corporea di origine sconosciuta, mancanza di appetito. Questi ed altri disturbi, una volta identificati, devono essere discussi con il pediatra e, se necessario, eseguita la diagnosi.
Se si sospetta un'anomalia congenita del sistema urinario, vengono prescritti un esame del sangue e un esame ecografico degli organi pelvici. Oltre all'esame del sangue, può essere prescritto anche un esame radiografico con l'introduzione di un mezzo di contrasto per determinare con precisione la posizione dell'anomalia.

Decorso della malattia
La stragrande maggioranza delle malformazioni congenite delle vie urinarie vengono diagnosticate accidentalmente (ad eccezione di quelle visualizzabili ad occhio nudo) durante l'esame del paziente per altri motivi; solitamente tali patologie non causano conseguenze negative in futuro, ma esistono una serie di casi di patologie gravi, ad esempio l'estrofia della vescica, che richiedono un trattamento complesso di emergenza.
La moderna terapia per le anomalie del sistema urinario è possibile solo se la patologia viene diagnosticata precocemente, quindi ogni bambino deve sottoporsi a tutti gli esami preventivi obbligatori.
Le malformazioni degli ureteri rappresentano il 22-25% di tutte le anomalie del sistema urinario e il 4,2-5% delle lesioni degli organi urinari. Alcune di queste anomalie vengono scoperte per caso. Altri possono causare una grave compromissione della funzionalità renale, favorendo la stasi urinaria e la fissazione dell’infezione nei reni.
Numerose anomalie nel numero, nella forma, nella posizione e nella struttura dell'uretere portano all'interruzione del deflusso dell'urina dal rene. L'urodinamica è disturbata non solo in presenza di ostacoli anatomicamente pronunciati, ma in quasi tutte le malformazioni delle vie urinarie superiori, anche con ostacoli impercettibili al deflusso dell'urina. Più spesso si osserva una dilatazione congenita o un restringimento dell'uretere.
Le anomalie degli ureteri sono spesso multiple, bilaterali e portano ad alterazioni del parenchima renale. Quanto più grave è l'anomalia, tanto prima si manifesta e viene diagnosticata.
Tutte le malformazioni degli ureteri portano a una compromissione della funzionalità renale, in particolare all'urodinamica. Eventuali disturbi nella formazione del germoglio ureterale si manifestano nella morfogenesi del parenchima renale, poiché un nefrone a tutti gli effetti può formarsi solo in condizioni di normale sviluppo sia del blastema metanefrogenico che delle parti distali dei dotti collettori, dei calici , bacino, ecc. Anomalie nella quantità dell'uretere e della pelvi renale si combinano con anomalie del parenchima renale.
Secondo la classificazione generalmente accettata, le anomalie ureterali sono suddivise nei seguenti gruppi:
anomalie quantitative - aplasia, raddoppiamento, triplicazione (completa o incompleta), ecc.;
anomalie strutturali - ipoplasia, restringimento (stenosi), valvola, diverticolo, ureterocele, displasia neuromuscolare, nonché acalasia, megauretere, idroureteronefrosi;
anomalie di forma - uretere a forma di anello, a cavatappi;
anomalie di localizzazione-uretere retrocavale, uretere retroiliaco, orifizio ureterale ectopico.
Le manifestazioni cliniche di varie anomalie ureterali dipendono non tanto dalle sue caratteristiche morfologiche, ma dalle complicanze causate dai disturbi dello sviluppo. Le principali complicanze sono malattie infiammatorie, idronefrosi, formazione di calcoli, ipertensione nefrogenica, che spesso si verificano sullo sfondo della pielonefrite. L'infezione dovuta ad anomalie dell'uretere si verifica più spesso nella prima infanzia. Il suo corso può essere attivo o con periodi di latenza, esacerbazioni periodiche. Più spesso, il processo infettivo è di natura ciclica a causa di malattie intercorrenti o di un aumento dello stress sul corpo (anni di crescita intensiva, pubertà, gravidanza). In rari casi, anche anomalie pronunciate non sono accompagnate da complicanze per lungo tempo e l'infezione compare solo in età adulta.
Anomalie dell'uretere spesso predeterminano lo sviluppo di idronefrosi o ureteroidronefrosi. A seconda del grado di pervietà dell'uretere e del livello di ostruzione, si osserva una perdita di funzione o la sua compensazione a causa dell'ipertrofia e dell'aumento della contrattilità dell'uretere nelle sezioni situate al di sopra del livello dell'anomalia. I pazienti spesso sviluppano idronefrosi progressiva, che porta all'insufficienza renale.
Se il deflusso dell'urina viene interrotto nelle parti inferiori delle vie urinarie, i processi di compensazione durano più a lungo e l'idronefrosi progredisce più lentamente. Un certo ruolo, oltre all'ipertrofia della parete ureterale oltre l'ostacolo, è giocato anche dall'aumento del campo di riassorbimento, che non si limita al reflusso a livello del rene, ma si manifesta in tutto l'uretere.
In caso di numerose anomalie dell'uretere le misure conservative sono molto raramente limitate. La minaccia di morte renale costringe a ricorrere a interventi chirurgici, le cui conseguenze dipendono dall'età del bambino. Quanto prima viene eseguita l'operazione, tanto maggiore è la speranza di un compenso funzionale dei reni, tanto più favorevole è la prognosi se combinato con il trattamento con agenti antibatterici, che consente di ottenere la remissione clinica e laboratoristica del processo infiammatorio solo nel 50% dei bambini. Ciò è spiegato dal fatto che la maggior parte delle anomalie, ad eccezione dei disturbi urodinamici, sono accompagnate da una formazione impropria del tessuto renale e dall'immaturità del sistema immunitario. Dopo il ripristino dell'urodinamica, questi cambiamenti rimangono la causa diretta del mantenimento del processo infiammatorio.
Nella diagnosi delle principali malformazioni degli ureteri, il ruolo principale è giocato dai metodi urologici - varianti dell'urografia escretoria. La pielografia ascendente è utilizzata relativamente raramente. Questo studio viene eseguito solo nei casi in cui l'anomalia ureterale provoca la completa perdita della funzionalità renale.
La cistoscopia è di particolare importanza poiché consente di determinare la posizione dell'orifizio ureterale, la sua contrattilità, la forma e la lunghezza della parte intravescicale. L'elevata incidenza di reflusso vescico-ureterale in tutte le anomalie ureterali rende possibile la sostituzione della pielografia ascendente con la cistografia minzionale convenzionale.
Informazioni importanti sono fornite da metodi basati sulle capacità dell'osservazione televisiva e della registrazione video della contrazione degli ureteri durante l'urografia escretoria. L'angiografia renale, i metodi di ricerca farmacodiagnostica, immuno-morfologica e cistochimica consentono di valutare lo stato morfo-funzionale del tessuto renale, delineare un piano di trattamento per il paziente, evitare errori diagnostici, correggere violazioni nell'operazione, monitorare l'ulteriore corso di la malattia e prevederne le possibili conseguenze.

ANOMALIE QUANTITATIVE
Aplasia (agenesia) dell'uretere È molto rara e rappresenta lo 0,2% delle anomalie renali e del tratto urinario. L'anomalia bilaterale è solitamente associata all'agenesia renale bilaterale, meno spesso al rene multicistico bilaterale. Questa anomalia non ha alcun significato clinico perché è incompatibile con la vita.
Anche l'aplasia ureterale unilaterale è una componente dell'aplasia renale ed è il risultato dell'assenza di un germoglio ureterale. A volte l'uretere si presenta sotto forma di un sottile cordone fibroso o di un processo che termina alla cieca.
La diagnosi di aplasia ureterale si basa sui dati dell'urografia escretoria, che consente di stabilire l'assenza di funzione di uno dei reni. La cistoscopia rivela ipoplasia o completa assenza della metà del triangolo vescicale. L'apertura dell'uretere può trovarsi nella sua posizione abituale, ma essere ristretta. Con l'osservazione a lungo termine è possibile rilevare l'assenza delle sue contrazioni. A volte il foro appare come una depressione cieca, che si determina al momento dell'inserimento del catetere, oppure termina alla cieca a qualsiasi livello. In questi casi, la cistografia è piuttosto istruttiva. In caso di apertura rudimentale dell'uretere, si consiglia di eseguire un esame ecografico e una tomografia computerizzata.
La necessità di trattamento nasce solo quando l'uretere termina alla cieca, poiché questa anomalia può provocare un processo infiammatorio, talvolta con suppurazione (empiema), e formazione di calcoli. In caso di cicatrici, l'apertura periferica dell'uretere forma una cavità chiusa, simile ad una cisti o ad un tumore della cavità addominale.
Tali complicazioni si manifestano con dolore nella corrispondente regione inguinale o epigastrica, disuria, aumento intermittente della temperatura corporea e fenomeni di intossicazione cronica. L'urina contiene un gran numero di leucociti, proteine e batteri. Se è presente un calcolo, viene rilevata macro o microematuria.
Il trattamento consiste nella rimozione del moncone ureterale.
Duplicazione dell'uretere - una delle anomalie più numerose (1:140). È causata dalla crescita simultanea di due ureteri da due germogli ureterali del blastema nefrogenico o dalla scissione di un singolo germoglio ureterale. Uno degli ureteri può svilupparsi normalmente, mentre l'altro può svilupparsi patologicamente. Se si formano più anlage ureterali nella sezione caudale del dotto del rene primario, è possibile non solo il raddoppio, ma anche il triplo degli ureteri morfologicamente completi. I due ureteri corrispondono a due pelvi renali, che sono collettori di urina per le diverse estremità del rene.
In questi casi, i reni vengono raramente isolati. Si forma un terzo germoglio aggiuntivo.
A volte due o più ureteri si estendono dalla pelvi di un rene non duplicato, oppure l’estremità prossimale di uno degli ureteri termina alla cieca. Entrambi gli ureteri solitamente passano attraverso la stessa guaina fasciale.
Si osserva la duplicazione completa (duplex dell'uretere) e incompleta (fisso dell'uretere) degli ureteri.
Con la duplicazione incompleta, entrambi gli ureteri si estendono dalla pelvi renale fino alla vescica e si fondono in uno solo a diverse distanze da essa. In questo caso, nella vescica appare un foro: un uretere diviso. A volte gli ureteri drenano vicino alla vescica, per via intravescicale (intramurale) o anche all'apertura. Uno degli ureteri confluisce nell'altro ad angolo acuto.
Di norma, la lunghezza di entrambi gli ureteri dal segmento ureteropelvico alla confluenza è diversa e le sezioni di entrambi gli ureteri sopra di esso si trovano in diverse fasi di peristalsi. Con il raddoppio incompleto, la scissione si osserva principalmente nel terzo superiore dell'uretere, meno spesso - nel mezzo e in 1/3 dei pazienti - in quello inferiore.
La duplicazione dell'uretere, sia completa che incompleta, è spesso unilaterale. Localizzato su entrambi i lati con la stessa frequenza.
Con la duplicazione completa, entrambi gli ureteri vanno separatamente alla vescica. Sono strettamente adiacenti alle pareti, secondo la legge di Weigert-Meyer, si intersecano nelle sezioni prossimale e distale e si aprono con due aperture sulla metà corrispondente del triangolo vescicale (una sopra l'altra o una accanto all'altra), se non c'è ectopia di uno di essi. Nella vescica, gli orifizi ureterali della pelvi superiore sono quasi sempre contenuti al di sotto degli orifizi ureterali della pelvi inferiore.
Il reflusso vescico-ureterale si osserva più spesso con la completa duplicazione degli ureteri. Ciò è dovuto alla breve sezione intravescicale dell'uretere, che si apre più prossimalmente. Occasionalmente si osserva una fine cieca di uno degli ureteri duplicati. L'anomalia si manifesta con dolore nella regione epigastrica e sintomi del processo infiammatorio.
La duplicazione degli ureteri è spesso associata ad altre malformazioni: assenza di entrambi o di uno degli orifizi ureterali, restringimento (stenosi ureterale, ureterocele, ectopia dell'apertura di uno degli ureteri (solitamente quello inferiore), displasia segmentale o diffusa dell'apparato neuromuscolare elementi degli ureteri, vasi aberranti, aderenze, cordoni fibrosi, ecc. .P..
Con un doppio uretere non si osservano sintomi caratteristici. Per molto tempo l'anomalia ha un decorso asintomatico. Le manifestazioni cliniche si verificano quando si verificano complicanze. I sintomi sono determinati dalla natura e dallo stadio della complicanza o dalle anomalie associate.
Il principale metodo diagnostico è l'urografia escretoria. Se la funzione di metà del doppio rene diminuisce, le informazioni necessarie possono essere ottenute mediante pielografia per infusione utilizzando immagini ritardate. Segni radiologici di duplicazione dell'uretere comprendono: assenza delle coppe superiori nella pelvi inferiore; spostamento della parte inferiore del bacino verso il basso, verso l'esterno o rotazione attorno all'asse longitudinale; deformazione delle coppe situate più in alto; la presenza di un'ampia zona “silente” tra il bordo del bacino e l'estremità del rene; la comparsa di reflusso tubulare pelvico-renale nell'area delle coppe superiori; la presenza di due pelvi renali o di due ureteri.
La cistoscopia può rilevare la duplicazione completa dell’uretere se entrambe le aperture si aprono nella vescica. La combinazione della cistoscopia con la somministrazione endovenosa di indaco carminio consente di determinare il valore funzionale dei reni superiori e inferiori. Utilizzando la cistografia minzionale seriale, viene rilevato il reflusso vescico-ureterale.
Trattamento. Se la duplicazione degli ureteri viene scoperta per caso e l'anomalia non provoca disturbi urodinamici evidenti, i bambini non necessitano di trattamento. Sono soggetti all'osservazione di un pediatra nel luogo di residenza. In caso di reclami è necessaria la consultazione. Nel caso in cui la duplicazione degli ureteri sia complicata dalla pielonefrite e l'esame uro-radiologico non riesca a rivelare disturbi urodinamici significativi, ai bambini dovrebbe essere prescritto un trattamento antinfiammatorio conservativo, una terapia stimolante e desensibilizzante e dovrebbero essere adottate misure per aumentare la reattività immunologica. Se tale trattamento è inefficace, si consiglia l’intervento chirurgico.
L'intervento chirurgico è necessario per complicazioni dell'anomalia come la formazione di calcoli, ureteroidronefrosi di una o entrambe le parti del doppio rene, grave reflusso vescico-ureterale e interureterale, ectopia di una delle aperture del doppio uretere, ureterocele.
La scelta del metodo di trattamento dipende da diversi fattori: il grado di danno al segmento renale, il tipo di duplicazione ureterale, la natura dei cambiamenti nella parte terminale dell'uretere, la presenza di reflusso nell'uretere adiacente e controlaterale.
L'intervento di scelta per l'orifizio ectopico dell'uretere accessorio, l'idro- e ureteronefrosclerosi segmentale, il reflusso vescico-ureterale e il piccolo ureterocele senza ostruzione e reflusso negli ureteri vicini è l'eminefroureterectomia. In caso di ureterocele vescicale o ectopico di grandi dimensioni, si rimuovono le sue membrane, eseguendo un'eminefrureterectomia e la correzione delle aperture degli ureteri adiacenti e controlaterali. Se le funzioni di entrambi i segmenti del rene sono preservate, sono applicabili operazioni di conservazione dell'organo: anastomosi degli ureteri nella cistoide superiore, operazioni antireflusso su due ureteri in un blocco.
Per eliminare il reflusso vescico-ureterale in caso di duplicazione degli ureteri si esegue un intervento di Politano-Leadbetter su uno o entrambi gli ureteri. È consigliabile integrare l'eminefrureterectomia con un intervento chirurgico antireflusso nei casi in cui uno dei segmenti renali ha perso completamente la sua capacità funzionale e l'uretere rimanente presenta un orifizio vescico-ureterale difettoso.
In caso di duplicazione completa e reflusso vescico-ureterale nel segmento inferiore del rene, l'ureterectomia deve essere utilizzata con due approcci: 1) nella parte bassa della schiena per rimuovere l'uretere e creare una pieloureteroanastomosi, 2) nella regione epigastrica per completare l'ureterectomia e immergere il moncone ureterale nella parete vescicale.
Triplicazione degli ureteri - un'anomalia molto rara. Sono stati descritti casi di presenza di 4, 6 e persino 12 ureteri.
Le manifestazioni cliniche, la diagnosi e il trattamento di questa anomalia sono gli stessi della duplicazione delle vie urinarie superiori. Nei casi non complicati, l'anomalia non viene diagnosticata. Clinicamente manifestato nel caso di un processo infiammatorio, ureteroidronefrosi o formazione di calcoli, nonché ectopia di una delle aperture dell'uretere.
ANOMALIE DI STRUTTURA E FORMA
Ipoplasia ureterale combinato con ipoplasia del rene corrispondente o della sua metà (con raddoppio), displasia renale multicistica. L'uretere ha l'aspetto di un tubo sottile con un diametro nettamente ridotto. Potrebbe essere cancellato in alcune aree. Più spesso il lume dell'uretere viene preservato. La sua parete è sottile a causa del sottosviluppo delle fibre muscolari, la peristalsi è fortemente indebolita.
Questa malformazione è spesso associata al reflusso vescico-ureterale. Le manifestazioni cliniche dell'ipoplasia ureterale sono causate da concomitanti anomalie renali.
La diagnosi dell'anomalia si basa sui dati dell'urografia escretoria e sulla sua modificazione dell'infusione, poiché l'uretere ipoplastico si estende dalla pelvi di un rene funzionante, la sua funzione è solitamente significativamente ridotta e un'immagine chiara delle vie urinarie può essere ottenuta solo con immagini ritardate .
La diagnosi definitiva può essere posta mediante pielografia retrograda, se è possibile l'inserimento di un catetere nell'uretere, oppure mediante cistografia, quando la malformazione è complicata da reflusso vescico-ureterale.
Trattamento dipende dalle condizioni del rene corrispondente. Quando si esegue una nefrectomia, viene rimosso anche l'uretere. L'obliterazione dell'uretere a diversi livelli o la sua conclusione cieca a causa di aplasia, le complicanze dovute al processo infiammatorio in una formazione simile a un diverticolo sono indicazioni per l'ureterectomia e la rimozione di un rene non funzionante.
Restringimento (stenosi) dell'uretere osservato nello 0,5-0,7% dei bambini. Molto spesso l'anomalia è localizzata nel segmento vescico-ureterale, quindi nel segmento ureteropelvico, ma può essere osservata in qualsiasi parte dell'uretere.
Il restringimento dell'uretere può essere unilaterale o bilaterale, singolo o multiplo. Nella maggior parte dei casi si tratta di una patologia congenita, ma può anche essere acquisita (in seguito a traumi, danni durante esami strumentali, piaghe da decubito o infiammazioni dovute alla presenza prolungata di un calcolo nell'uretere). Dopo l’intervento chirurgico sull’uretere può svilupparsi un restringimento. Se non esiste una storia di nessuno di questi motivi, possiamo supporre che il restringimento sia congenito. I restringimenti congeniti e acquisiti sono clinicamente e istologicamente indistinguibili.
Al di sopra del restringimento dell'uretere e del sistema collettore si espande a causa del costante aumento della pressione e del ristagno di urina. Se l'ostruzione è localizzata nel segmento ureteropelvico si sviluppa idronefrosi. Quando il restringimento è localizzato nella parte prostatica (iuxtavescicale) o nel terzo medio dell'uretere, si espande notevolmente e si allunga al di sopra dell'ostruzione. L'uretere diventa lungo, tortuoso e può essere spesso quanto il colon. Nei bambini piccoli con struttura corporea astenica e tono muscolare ridotto, tali ureteri possono essere palpati attraverso la parete addominale anteriore. Questa patologia viene spesso confusa con il megauretere.
Il quadro clinico è causato dalla ridotta pervietà dell'uretere, dallo sviluppo di idronefrosi o ureteroidronefrosi e dall'aggiunta di infezione.
Il metodo principale per diagnosticare il restringimento dell'uretere è l'urografia escretoria.
Il trattamento è chirurgico, la sua portata dipende dalle condizioni del tratto urinario superiore e dalla posizione del restringimento. Consiste nell'asportazione della parte ristretta dell'uretere e nel ripristino della sua pervietà, nel trapianto dell'uretere nella vescica, nella sostituzione parziale o completa con un segmento dell'intestino. Se il parenchima renale viene distrutto, è indicata la nefroureterectomia.
Valvola ureterale è una piega trasversale congenita della mucosa contenente fibre muscolari circolari.
Fino alla comparsa dell'ureteroidronefrosi e all'aggiunta dell'infezione, l'anomalia non si manifesta clinicamente. Il processo infiammatorio nei reni colpiti e controlaterali è precocemente complicato da un'ostruzione (ostruzione) a qualsiasi livello dell'uretere. Ciò è spiegato dall'aumento della pressione nella pelvi renale, dalla stasi venosa e linfatica e dall'ischemia del parenchima renale.
La diagnosi viene stabilita sulla base dell'urografia escretoria e della pielografia retrograda. L'arteriografia renale consente di determinare lo stadio dell'idronefrosi, ma non determina la causa dell'ostruzione nell'uretere. I metodi con radionuclidi (scansione, renografia) forniscono informazioni sulla quantità di parenchima renale rimanente. Spesso la tipologia dell'anomalia viene chiarita solo in corso d'intervento. Se l'idronefrosi provoca una forte espansione delle cavità renali e la morte quasi completa del suo parenchima, viene utilizzata la pielografia con puntura percutanea anterograda per differenziarla dal danno renale cistico unilaterale.
Il trattamento consiste nella rimozione (resezione) dell'area interessata e nel ripristino della pervietà dell'uretere suturandone le estremità. Se la valvola viene posizionata nella parte pelvica dell'uretere, dopo la sua rimozione, secondo le indicazioni, l'uretere viene trapiantato nella vescica o la sua chirurgia plastica viene eseguita utilizzando il metodo Boari.
La nefrectomia e l'ureterectomia per il restringimento e la valvola ureterale vengono eseguite solo quando il parenchima renale si è trasformato in una sacca a pareti sottili o quando l'idronefrosi a seguito di infezione è stata complicata dalla pionefrosi. In altri casi, le indicazioni per la nefrectomia dovrebbero essere nettamente limitate.
Diverticolo ureterale Come patologia congenita, si osserva molto raramente. Questa è una lunga formazione sferica che si collega al lume dell'uretere. Le sue pareti hanno tutti gli stessi strati delle pareti dell'uretere. Il diverticolo si sviluppa come un'ulteriore gemma ureterale. Nella maggior parte dei casi, si forma nel terzo inferiore dell'uretere. È più spesso localizzato a sinistra e molto raramente su entrambi i lati.
Il diverticolo ureterale congenito è solitamente piccolo e non causa alcun problema. A volte raggiunge grandi dimensioni, comprime l'uretere, causando ureteroidronefrosi e, se infetto, pionefrosi. Un sintomo comune è la piuria persistente.
Il metodo principale per diagnosticare questa anomalia è la pielografia escretoria e retrograda. Sugli ureteropielogrammi nel terzo inferiore dell'uretere è presente una notevole formazione sferica, un'estremità della quale termina cieca e la seconda è combinata con l'uretere. A volte questa formazione si trova vicino al rene e quindi viene scambiata per un uretere aggiuntivo. In questi casi, l'urografia escretoria, l'ecografia e la risonanza magnetica aiutano a chiarire la diagnosi. Negli urogrammi, il diverticolo è riempito con mezzo di contrasto radiografico solo nella sua parte inferiore, e talvolta non è riempito affatto.
Trattamento consiste nell'asportazione del diverticolo. Per l'ureteropionefrosi, la nefroureterectomia è efficace.
Ureterocele - protrusione simile a una cisti nella vescica della mucosa o in tutti gli strati della parete della parte intravescicale dell'uretere. Si osserva più spesso a destra, spesso su entrambi i lati. La dimensione dell'ureterocele varia da pochi millimetri a diversi centimetri di diametro. A volte raggiunge grandi dimensioni, riempie gran parte della vescica e nelle donne può fuoriuscire attraverso l'uretra, simulando i segni della vescica.
La causa dell'ureterocele è un restringimento dell'apertura dell'uretere o un cambiamento nella sua parte intravescicale. L'urina viene trattenuta nell'uretere sopra il restringimento, rendendone difficile la rimozione. La mucosa del bordo inferiore dell'uretere, debolmente collegata alla parete muscolare, scivola nella cavità vescicale sotto la pressione dell'urina e delle onde peristaltiche, creando sporgenze a forma di sacco. Consiste di due strati di mucosa: all'interno - la mucosa ureterale, all'esterno - la mucosa della vescica. Spesso nella sua cavità si formano delle pietre.
Si distinguono i seguenti tipi di ureterocele: 1) semplice; 2) che sporge nell'uretra (nelle donne); 3) che, a causa dell'ectopia, si apre nell'uretra, diverticolo della vescica; 4) che termina alla cieca.
Un ureterocele piccolo non ha manifestazioni cliniche, ma uno grande può causare disuria e persino ritenzione urinaria completa. Con l'esistenza prolungata dell'ureterocele, si sviluppa la dilatazione dell'uretere e della pelvi renale con la formazione di ureteroidronefrosi e persino pioiefrosi. In questi casi, la funzionalità renale è compromessa.
Il metodo principale per diagnosticare l'ureterocele è la cistoscopia. Quando si esamina il sito dell'apertura ureterale, viene rilevata una sporgenza, che diminuisce o aumenta. Sui cistogrammi si osserva un difetto di riempimento a forma di semianello, che copre la cavità con liquido radiopaco. L'urografia escretoria consente di chiarire le condizioni delle vie urinarie superiori e identificare i cambiamenti caratteristici. L’ecografia è ampiamente utilizzata per diagnosticare l’ureterocele. Negli ecogrammi, un ureterocele è una formazione occupante spazio piena di liquido nella proiezione della vescica, delimitata da essa da una membrana sottile e ben definita. Molti pazienti affetti da ureterocele sviluppano ureteroidronefrosi. Questo può essere determinato utilizzando gli ultrasuoni e la tomografia computerizzata.
Trattamento. Viene eseguita la dissezione transuretrale dell'ureterocele. Se l'ureterocele è grande o sporge dalla vescica attraverso l'uretra, viene asportato transvescicamente e la mucosa vescicale viene suturata alla mucosa ureterale. Quando si asporta un ureterocele, l'apparato di chiusura (sfintere) dell'uretere viene spesso interrotto, il che porta al reflusso e all'ureteroidronefrosi.
Se il parenchima renale muore, viene eseguita la nefroureterectomia.
N ehm chiaramente -displasia muscolare dell'uretere - restringimento congenito bilaterale dell'apertura e della parte intravescicale dell'uretere in combinazione con displasia neuromuscolare del suo terzo inferiore, che porta alla dilatazione dell'uretere - megauretere .
Esistono due punti di vista riguardo all'eziologia e alla patogenesi di questa anomalia: I) insufficienza di sviluppo dell'apparato neuromuscolare dell'uretere, 2) presenza di ostacoli funzionali o organici a livello della prostata o della parte intravescicale dell'uretere.
Esistono due forme di questa anomalia: 1) causata da un'ostruzione meccanica nella parte prostatica o intravescicale dell'uretere, che impedisce il deflusso dell'urina dalla cistoide pelvica, 2) associata a un disturbo meccanico o funzionale dell'urodinamica della zona dell'apertura ureterale, del collo della vescica o dell'uretra. La differenza tra loro è che alla seconda forma si associa il reflusso vescico-ureterale, che è la causa diretta della dilatazione dell'uretere.
Primo stadio della displasia neuromuscolare dell'uretere (acalasia) caratterizzato dall'espansione del suo cistoide inferiore, il secondo (megaumetro) - espansione dell'uretere su tutta la sua lunghezza; terzo (idroureterone e congelato) - completa interruzione irreversibile dell'urodinamica e trasformazione idronefrotica del rene.
Clinicamente l'anomalia non si manifesta. I suoi sintomi si osservano se il ristagno di urina porta a gravi complicazioni: ureteroidronefrosi, pielonefrite, pionefrosi, ecc.
L'aggiunta di un processo infettivo porta alla comparsa di sintomi di pielonefrite. Inoltre, con il progredire dei disturbi urodinamici e della distruzione renale dovuta a pielonefrite e idronefrosi, compaiono segni di insufficienza renale cronica. I bambini crescono lentamente, i parametri biochimici del sangue cambiano. Pertanto, i sintomi di piuria cronica nei bambini, che si ripresentano, costituiscono un'indicazione per uno speciale esame urologico.
Il metodo principale per diagnosticare le anomalie è l'esame radiografico. Durante la cistografia, a causa della contrazione incompleta dell'apertura ureterale, si può notare il riempimento di un uretere fortemente dilatato, spesso con piegature dovute al reflusso vescico-ureterale. I dati dell'urografia escretoria consentono di valutare la funzionalità renale. Usando l'urogramma, puoi tracciare il corso dell'uretere e determinare il grado della sua espansione. All'ureteropielogramma è chiaramente visibile un uretere tortuoso e fortemente dilatato con un'ectopia relativamente lieve della pelvi renale. Lo studio urochimografico sullo sfondo del reflusso ci consente di giudicare la peristalsi dell'uretere.
Il trattamento è solo chirurgico. Se l'espansione è limitata al segmento finale dell'uretere, allora si supera l'ostacolo e si esegue il reimpianto nella vescica. Alcuni autori integrano il reimpianto con la rimozione della maggior parte dell'uretere dilatato.
I metodi di trattamento chirurgico possono essere suddivisi nei seguenti gruppi: 1) ureterocistoneostomia con formazione di una “proboscide” intravescicale; 2) ureteroneocistostomia con tunnel sottomucoso; 3) allungamento della sezione intravescicale dell'uretere mantenendone l'apertura; 4) allungamento della sezione intravescicale dell'uretere dovuto allo spostamento o alla chirurgia plastica della sua apertura; 5) creazione di un duplicato dell'uretere dilatato, ureterocistoanastomosi antireflusso; 6) protesi ureterale.
La lotta contro le infezioni e i disturbi dell’omeostasi è di grande importanza. La terapia antibatterica viene utilizzata prima di eseguire un intervento di chirurgia plastica, nel periodo postoperatorio allo scopo di igienizzare i reni e le vie urinarie e in assenza di indicazioni per l'intervento chirurgico - come metodo indipendente.
Gli interventi sull'uretere sono complessi e non sempre efficaci, soprattutto nelle fasi successive della malattia.
Uretere anulare - una rara anomalia in cui l'uretere nel terzo medio è attorcigliato a forma di anello. L'embriogenesi di questa anomalia è probabilmente legata all'incapacità dell'uretere pelvico di ruotare insieme al rene. Questa malformazione non causa un forte disturbo dell'urodinamica, quindi le sue manifestazioni cliniche sono scarse. Se si verifica un'infiammazione, la curva dell'uretere si fissa e i disturbi nel deflusso dell'urina iniziano a manifestarsi sotto forma di colica renale. La patologia può essere complicata dalla formazione di calcoli e ureteroidronefrosi.
Per diagnosticare questa anomalia si utilizza l'urografia escretoria con immagini ritardate. In rari casi, è necessaria una pielografia retrograda.
Trattamento. Se un uretere anulare causa stasi urinaria, la resezione ureterale viene eseguita con anastomosi termino-terminale.
ANOMALIE DI UBICAZIONE
Uretere retrocavale - il risultato di un'anomalia della vena cava inferiore, in cui l'uretere nel terzo superiore copre a spirale la vena cava inferiore e, partendo dal terzo medio, va nella solita direzione - nella vescica. Questa posizione insolita dell'uretere porta ad un deflusso alterato dell'urina e alla trasformazione idronefrotica.
Lo sviluppo dell'idronefrosi e dell'infezione dell'uretere retrocavale non ha manifestazioni cliniche e viene diagnosticato per caso. La piuria persistente è spesso causata da pielonefrite cronica.
La diagnosi viene stabilita sulla base dei dati dell'urografia escretoria e, se la funzionalità renale diminuisce, della pielografia retrograda. Per chiarire la diagnosi, la cavografia viene talvolta utilizzata in combinazione con l'ureteropielografia.
Il trattamento di questa anomalia è chirurgico: intersezione e spostamento in avanti dell'uretere dalla vena cava con ripristino della sua pervietà. Se il parenchima renale muore, viene eseguita la nefroureterectomia.
Uretere retroiliaco - un'anomalia molto rara. Caratterizzato dalla posizione dell'uretere dietro l'arteria iliaca comune.
Clinica. La diagnosi e il trattamento di questa anomalia sono gli stessi dell'uretere retrocavale.
E fotocopia dell'orifizio ureterale - la posizione di una o entrambe le aperture degli ureteri in un luogo atipico. L'apertura ectopica di solito appartiene a uno degli ureteri duplicati. Nella maggior parte dei casi, l’uretere, che drena la pelvi renale superiore, si apre a un livello inferiore. Nelle ragazze, l'apertura dell'uretere ectopico si apre solitamente nella volta vaginale o in altre parti dei genitali esterni, nell'uretra vicino alla sua apertura esterna, nel retto, nella cervice o nel corpo dell'utero; nei ragazzi nella parte posteriore dell'uretra, dotti deferenti, vescicole seminali, perineo, retto, ecc.
L'urografia escretoria o infusionale, la cistoscopia, l'uretero e la colpografia aiutano a stabilire correttamente la diagnosi. Informazioni indirette sulla localizzazione dell'apertura ectopica dell'uretere possono essere fornite dalla pielografia retrograda eseguita attraverso un'apertura normalmente localizzata. L'ottenimento di una pielogramma retrogrado caratteristico di una doppia pelvi renale (un'immagine della pelvi principale sul lato dell'anomalia, leggermente più in basso e laterale rispetto al lato opposto) consente di determinare con alta probabilità il lato dell'orifizio ureterale ectopico .
In assenza di un'anomalia renale, la localizzazione dell'apertura ectopica è determinata sulla base di una forte diminuzione della funzionalità renale (secondo i dati dell'urografia e della scansione per infusione) e dell'assenza (durante la cistoscopia) nella vescica su un lato del l'apertura ureterale.
Se si sospetta la possibilità di localizzare l'apertura dell'uretere nel tratto di efflusso familiare, deve essere utilizzata la vescicolografia.
Il trattamento dell’orifizio ureterale ectopico è chirurgico. In presenza di un'ampia apertura senza concomitante duplicazione del tratto urinario superiore, è indicata l'ureterocistoneostomia. Se la funzione del doppio rene viene preservata, viene creata un'ureterouretroanastomosi o ureteropieloanastomosi. Con una forte diminuzione della funzione della metà corrispondente del rene, l'eminefrectomia con ureterectomia è efficace. Se il rene non funziona, viene eseguita la nefroureterectomia. La rimozione di un rene in caso di orifizio ureterale ectopico viene effettuata rigorosamente secondo le indicazioni.
Reflusso vescico-ureterale può essere dovuto ad insufficienza congenita dell'apparato otturatore dell'orifizio ureterale o ad alterata pervietà del segmento vescico-ureterale e ad un processo infiammatorio cronico.
Esistono opinioni diametralmente opposte riguardo alla connessione del reflusso con anomalie del segmento vescico-ureterale. Alcuni autori ritengono che il reflusso si verifichi a causa della ritenzione urinaria cronica e dell'infiammazione. Altri - che è causato da una violazione del normale stato anatomico e fisiologico del segmento vescico-ureterale. La frequenza del reflusso con duplicazione degli ureteri indica che la sua comparsa è dovuta a uno e ad altri motivi.
Quindi il reflusso vescico-ureterale deve essere considerato come una condizione al limite delle anomalie dell'uretere e della vescica. La chiusura dell'apertura dell'uretere è assicurata da un aumento della pressione intravescicale dovuto a due fattori: a) la direzione obliqua dell'uretere nel suo passaggio attraverso la parete muscolare della vescica e la compressione dell'uretere durante la contrazione del muscolo che spinge urina, b) la posizione sottomucosa della parte finale, intravescicale, dell'uretere, per circa 11 mm. Quando la pressione nella vescica aumenta, questa sezione dell'uretere viene premuta contro la parete muscolare.
La chiusura dell'apertura ureterale viene interrotta quando la parete muscolare si trova in una direzione perpendicolare o vicina ad essa, nonché quando la parte sottomucosa (intravescicale) viene accorciata. Questa posizione si crea con il completo sdoppiamento dell'uretere, ma può verificarsi senza questa anomalia. Nei bambini con una parte sottomucosa corta dell'uretere, la maturazione può avvenire prima della pubertà con la scomparsa del reflusso. Tuttavia, ciò può essere prevenuto dall'infezione e dalla progressiva ostruzione del segmento vescico-ureterale, come evidenziato dalla scomparsa del reflusso quando l'infezione è sotto controllo. In effetti si crea un circolo vizioso: il reflusso favorisce un'infezione delle vie urinarie ascendenti e l'infezione mantiene il reflusso. Questo cerchio può essere spezzato solo con una terapia antibatterica mirata con la somministrazione di grandi quantità di liquidi.
La diagnosi viene effettuata sulla base dei risultati della cistografia e dei metodi di ricerca sui radionuclidi. Assicurati di determinare le condizioni del segmento vescico-ureterale. Se la sua pervietà è compromessa, il reflusso può essere eliminato solo dopo la correzione di detta anomalia.
A seconda della gravità del reflusso del fluido radiopaco e della dilatazione delle vie urinarie superiori, sull'urogramma si possono rintracciare cinque gradi di reflusso vescico-ureterale: I - reflusso della sostanza radiopaca nella parte distale dell'uretere; II-riempimento dell'uretere e del sistema calice-pelvi con una sostanza radiopaca; III - moderata dilatazione dell'uretere con pieloectasia e arrotondamento delle arcate del calice; IV - pronunciata espansione e tortuosità dell'uretere, deformazione del sistema pielocaliceale; V - idrouretere e forte assottigliamento del parenchima renale.
Trattamento. Nella maggior parte dei casi, il reflusso vescico-ureterale nei bambini scompare con l’età. Ciò si spiega con il completamento dello sviluppo della sezione corrispondente dell'uretere grazie al raggiungimento di una lunghezza sufficiente nello strato sottomucoso della vescica. Pertanto, se l'inferiorità congenita dell'apparato otturatorio dell'uretere è moderata (I, II, talvolta III grado) e il processo corrispondente non è ancora iniziato o è benigno, è consigliabile effettuare una terapia conservativa (uroantisettici, elettrodo esterno stimolazione della vescica con correnti sinusoidali o diadinamiche). Se è inefficace, vengono eseguite operazioni antireflusso. Prima di ciò, dovrebbero essere eliminate le cause dell'ostruzione del collo della vescica e dell'uretra, se presenti. Negli stadi IV e V si raccomanda la resezione dell'uretere distale con ureterocistoanastomosi.
I bambini che hanno subito un intervento chirurgico per reflusso vescico-ureterale sono soggetti alla supervisione di un urologo e di un nefrologo.
5510 0
Malattia del rene policistico di tipo infantile(malattia del rene policistico di tipo I sindrome di Potter, malattia del rene policistico autosomica recessiva) è un ingrossamento simmetrico bilaterale dei reni dovuto alla sostituzione del parenchima con dotti collettori secondariamente dilatati senza proliferazione del tessuto connettivo.
La malattia si verifica con una frequenza di 2:110.000 nati ed è trasmessa con modalità autosomica recessiva. Si basa su un difetto primario dei dotti collettori. La pelvi renale, i calici e le papille rimangono intatti, poiché non vi è alcun difetto nella gemma ureterale. Le anomalie combinate sono rare.
Diagnostica: L'esame ecografico rivela un ingrossamento bilaterale dei reni sullo sfondo dell'oligoidramnios e l'assenza di ecotenosi della vescica. La tipica iperecogenicità renale è accompagnata da un aumento della conduttività del suono dovuta a molteplici strutture cistiche microscopiche nel parenchima renale.
Previsione sfavorevole. La morte dei neonati avviene per insufficienza renale.
Tattiche ostetriche: interruzione della gravidanza in qualsiasi momento.
Malattia del rene policistico di tipo adulto(malattia autosomica dominante, malattia policistica epatorenale dell'adulto, sindrome di Potter di tipo III) è caratterizzata dalla sostituzione del parenchima renale con numerose cisti di diverse dimensioni, che si formano a causa dell'espansione dei dotti collettori e di altri segmenti tubulari del nefrone. Le cisti coesistono con il tessuto renale intatto. I reni sono colpiti su entrambi i lati e risultano ingrossati, ma la prima manifestazione della malattia può essere un processo unilaterale. Il coinvolgimento del fegato nel processo è osservato meno frequentemente e si esprime nella fibrosi periportale, che è di natura focale.
Eziologia sconosciuto. Il tipo di ereditarietà comporta un rischio del 50% di sviluppare la malattia. Il suo focus genetico si trova sulla 16a coppia di cromosomi.
Frequenza: una persona su 1000 è portatrice del gene mutante; la penetrazione del gene avviene nel 100% dei casi, ma la sua espressività può variare da forme gravi che portano alla morte nel periodo neonatale a forme asintomatiche rilevate all'autopsia.
Clinicamente, il tipo policistico dell'adulto è combinato con alterazioni cistiche nel fegato, nel pancreas, nella milza, nei polmoni, nelle ovaie e nell'epididimo.
La diagnosi prenatale può essere effettuata mediante prelievo di villi coriali ed esame della biopsia utilizzando una speciale sonda genetica per individuare il locus del gene mutante. Vengono descritte singole osservazioni della diagnostica ecografica di questa patologia.
Previsione: la malattia del rene policistico è una malattia cronica; i suoi primi sintomi possono comparire a qualsiasi età. L'età media di insorgenza delle manifestazioni cliniche (dolore lombare, reni ingrossati, insufficienza renale e uremia) è di 35 anni. L'ipertensione è registrata nel 50-70% dei pazienti.
Tattiche ostetriche: i genitori a rischio dovrebbero essere informati sulla possibilità di diagnosticare la patologia nel primo trimestre di gravidanza e di interrompere la gravidanza nelle fasi iniziali; Una volta che il feto raggiunge la vitalità, vengono generalmente accettate le tattiche ostetriche standard.
Malattia renale multicistica(malattia renale multicistica, malattia renale cistica di tipo II, sindrome di Potter, malattia renale displastica) è un'anomalia renale congenita che si manifesta con degenerazione cistica del parenchima renale dovuta alla dilatazione primaria dei tubuli renali. Il processo può essere bilaterale, unilaterale o segmentale.
La malattia si manifesta principalmente in modo sporadico e può essere una manifestazione secondaria nell'ambito di altre sindromi:
- autosomico recessivo (sindrome di Meckel, sindrome di Dandy-Walker, sindrome di Roberts, sindrome di Smith-Lemli-Opitz, ecc.);
- autosomico dominante (sindrome di Apert);
- disturbi cromosomici.
Un processo bilaterale si verifica in uno su 10.000 neonati. Un processo unilaterale si osserva 2 volte più spesso nei neonati maschi.
I meccanismi patogenetici della malattia non sono noti. Si ritiene che questa complessa anomalia possa essere la conseguenza di due tipi di danno: il fallimento del blastema metanefrico, responsabile della formazione dei nefroni, e l'uropatia ostruttiva precoce.
Le lesioni unilaterali possono essere associate a difetti del tubo neurale, ernia diaframmatica, palatoschisi e anomalie gastrointestinali.
La diagnosi prenatale si basa sul rilevamento di reni cistici mediante ecografia, visualizzazione insufficiente della vescica, anche dopo un test con furosemide.
Previsione: un processo bilaterale, nella maggior parte dei casi, porta alla morte.
Tattiche ostetriche: in caso di processo bilaterale diagnosticato nelle prime fasi della gravidanza, si consiglia l'interruzione della gravidanza; un processo unilaterale con un cariotipo normale e senza anomalie associate non influisce sulla tattica ostetrica. Si consiglia che il parto avvenga in un centro perinatale.
Idronefrosiè il risultato dell'ostruzione delle vie urinarie alla giunzione della pelvi renale e dell'uretere.
La sua frequenza non è stata stabilita, poiché l'anomalia è un fenomeno sporadico. Dopo la nascita, viene diagnosticata 5 volte più spesso nei maschi.
Nei bambini con ostruzione della giunzione ureteropelvica, nel 27% dei casi si riscontrano anomalie combinate delle vie urinarie, tra cui reflusso vescico-ureterale, duplicazione bilaterale degli ureteri, megauretere ostruttivo bilaterale, rene controlaterale non funzionante, agenesia del rene controlaterale. Nel 19% si notano anomalie di altri organi e sistemi.
L'ostruzione della giunzione ureteropelvica viene diagnosticata mediante ecografia, basata sul rilevamento della dilatazione della pelvi renale.
Con un processo unilaterale, la prognosi è favorevole. Le tattiche ostetriche dipendono dal tempo di rilevamento del processo patologico e dal grado di disfunzione renale.
Lezioni selezionate di ostetricia e ginecologia
Ed. UN. Strizhakova, A.I. Davydova, L.D. Belotserkovtseva