Trombocitopenia immune nei bambini. Classificazione fisiopatologica della trombocitopenia Criteri diagnostici per la porpora trombocitopenica
107. Porpora trombocitopenica nei bambini. Eziologia, patogenesi, classificazione, quadro clinico, trattamento.
La porpora trombocitopenica idiopatica (autoimmune) è una malattia caratterizzata da una diminuzione isolata del numero di piastrine (meno di 100.000/mm3) con un numero normale o aumentato di megacariociti nel midollo osseo e dalla presenza di anticorpi antipiastrinici sulla superficie delle piastrine e nel siero del sangue, causando una maggiore distruzione delle piastrine.
Prevalenza, fattori di rischio ed eziologia. La frequenza della porpora trombocitopenica idiopatica nei bambini è di circa 1,5-2 ogni 100.000 bambini, senza differenze per genere, con pari frequenza delle forme acute e croniche. Nell'adolescenza, il numero delle ragazze malate diventa il doppio di quello dei ragazzi.
La causa della porpora trombocitopenica non è stabilita con precisione; Tra i fattori che precedono lo sviluppo della porpora trombocitopenica idiopatica vi sono infezioni virali e batteriche (40% dei casi), vaccinazioni e somministrazione di gammaglobuline (5,5%), interventi chirurgici e lesioni (6%); nel 45% dei casi la malattia si manifesta spontaneamente senza cause precedenti. Nella maggior parte dei pazienti con porpora trombocitopenica idiopatica, il background premorboso, lo sviluppo fisico e psicomotorio non differiscono da quelli dei bambini sani.
Il termine “idiopatico” indica un'insorgenza spontanea della malattia e un'eziologia finora non identificata.
Patogenesi della porpora trombocitopenica. La trombocitopenia porta all'interruzione dell'emostasi piastrinica e contribuisce allo sviluppo della sindrome emorragica di tipo petecchiale (microcircolatoria). La trombocitopenia è accompagnata da insufficienza angiotrofica, che provoca cambiamenti distrofici nell'endotelio dei piccoli vasi e dei capillari e porta ad una diminuzione della resistenza della parete vascolare e ad un aumento della sua porosità per i globuli rossi. Ciò si manifesta con emorragie puntiformi (petecchie) nelle zone di maggiore pressione idrostatica (arti inferiori); il numero delle petecchie può essere facilmente aumentato mediante la compressione degli arti con un laccio emostatico.
La sindrome emorragica nella porpora trombocitopenica idiopatica è caratterizzata da sanguinamento prolungato dai piccoli vasi, dovuto all'incapacità delle piastrine di formare un tappo piastrinico nei siti di danno endoteliale. Cambiamenti significativi si verificano nella parete vascolare e sotto l'influenza del processo patoimmune. A causa della comunanza delle strutture antigeniche delle piastrine e delle cellule endoteliali, le cellule endoteliali vengono distrutte dagli anticorpi antipiastrinici, il che aumenta le manifestazioni cliniche della sindrome emorragica.
Nella patogenesi della porpora trombocitopenica idiopatica, di fondamentale importanza è la sintesi immunopatologica di autoanticorpi antipiastrinici (IgG) da parte dei linfociti della milza, che si fissano su vari recettori di membrana delle piastrine e dei megacariociti, il che conferma la natura patoimmune della malattia e l'ipotesi di disfunzione primaria del sistema linfoide nella porpora trombocitopenica idiopatica. Come risultato del processo autoimmune, le piastrine perdono le loro proprietà di aggregazione adesiva e muoiono rapidamente, venendo assorbite dalle cellule mononucleari della milza e, nei casi più gravi, del fegato e di altri organi del sistema reticoloendoteliale (tipo “diffuso” di piastrine). sequestro). Con il sequestro piastrinico di tipo “diffuso”, la splenectomia non è sufficientemente efficace. Il tempo di dimezzamento della loro scomparsa è di mezz'ora o meno.
Nella porpora trombocitopenica idiopatica, sebbene il numero di megacariociti nel midollo osseo aumenti in modo significativo, sono caratterizzati da immaturità funzionale (il numero di forme immature aumenta e il numero di quelle funzionalmente attive diminuisce).
La porpora trombocitopenica idiopatica (autoimmune) può essere acuta, cronica e ricorrente. Nella forma acuta, la conta piastrinica si normalizza (più di 150.000/mm3) entro 6 mesi dalla diagnosi senza recidiva. Nella forma cronica, la trombocitopenia inferiore a 150.000/mm3 dura più di 6 mesi. Nella forma ricorrente, la conta piastrinica diminuisce nuovamente dopo essere tornata a livelli normali. La forma acuta è più tipica per i bambini e la forma cronica per gli adulti.
A causa del fatto che la porpora trombocitopenica idiopatica spesso si manifesta in modo transitorio, la reale incidenza non è stata stabilita. L'incidenza riportata è di circa 1 ogni 10.000 casi all'anno (3-4 ogni 10.000 casi all'anno tra i bambini sotto i 15 anni di età).
Come accennato in precedenza, la patogenesi della porpora trombocitopenica idiopatica si basa sull'aumento della distruzione delle piastrine cariche di autoanticorpi da parte delle cellule del sistema reticoloistiocitario. Negli esperimenti con piastrine marcate, si è riscontrato che la durata della vita delle piastrine diminuisce da 1-4 ore a diversi minuti. L'aumento del contenuto di immunoglobuline (IgG) sulla superficie delle piastrine e la frequenza della distruzione piastrinica nella porpora trombocitopenica idiopatica sono proporzionali al livello di IgG associate alle piastrine (PAIgG). I bersagli degli autoanticorpi sono le glicoproteine di membrana piastrinica (Gp): Gp Ilb/IIIa, Gp Ib/IX e Gp V.
Le persone con fenotipo HLA B8 e B12 hanno un rischio maggiore di sviluppare la malattia se presentano fattori precipitanti (complessi antigene-anticorpo).
Il picco di incidenza della porpora trombocitopenica idiopatica si verifica tra i 2 e gli 8 anni, con la stessa frequenza nei ragazzi e nelle ragazze. Nei bambini di età inferiore a 2 anni (forma infantile), la malattia è caratterizzata da un esordio acuto, un decorso clinico grave con sviluppo di trombocitopenia profonda inferiore a 20.000/mm3, una scarsa risposta all'esposizione e frequente cronicità del processo - fino al 30% dei casi. Il rischio di insorgenza di porpora trombocitopenica cronica idiopatica nei bambini è aumentato anche nelle ragazze di età superiore a 10 anni con una durata della malattia superiore a 2-4 settimane prima della diagnosi e una conta piastrinica superiore a 50.000/mm3.
Nel 50-80% dei casi, la malattia si manifesta 2-3 settimane dopo una malattia infettiva o un'immunizzazione (vaiolo, vaccino vivo contro il morbillo, ecc.). Molto spesso, l'insorgenza della porpora trombocitopenica idiopatica è associata a infezioni non specifiche del tratto respiratorio superiore, in circa il 20% dei casi - specifiche (morbillo, rosolia, morbillo, varicella, pertosse, parotite, mononucleosi infettiva, infezioni batteriche).
I sintomi della porpora trombocitopenica idiopatica dipendono dalla gravità della trombocitopenia. La sindrome emorragica si manifesta sotto forma di molteplici lividi petecchiali sulla pelle ed emorragie sulle mucose. Poiché petecchie (1-2 mm), porpora (2-5 mm) ed ecchimosi (più di 5 mm) possono accompagnare anche altre condizioni emorragiche, la diagnosi differenziale si basa sul numero di piastrine nel sangue periferico e sulla durata del sanguinamento. .
Il sanguinamento si verifica quando la conta piastrinica scende al di sotto di 50.000/mm3. Il rischio di sanguinamento grave si verifica con trombocitopenia profonda inferiore a 30.000/mm3. All'esordio della malattia, il sanguinamento nasale, gengivale, gastrointestinale e renale è solitamente insolito, così come il vomito di fondi di caffè e melena è raro. È possibile un grave sanguinamento uterino. Nel 50% dei casi, la malattia si manifesta con la tendenza a formare ecchimosi in luoghi di lividi, sulla superficie anteriore degli arti inferiori, sopra le sporgenze ossee. Anche gli ematomi muscolari profondi e l'emartro non sono tipici, ma possono essere il risultato di iniezioni intramuscolari e traumi estesi. Con la trombocitopenia profonda, si verificano emorragie nella retina dell'occhio e, raramente, sanguinamento nell'orecchio medio, che porta alla perdita dell'udito. L'emorragia cerebrale si verifica nell'1% dei casi con porpora trombocitopenica idiopatica acuta, nel 3-5% dei casi con porpora trombocitopenica idiopatica cronica. Di solito è preceduto da mal di testa, vertigini e sanguinamento acuto in qualche altra sede.
Un esame obiettivo può rivelare la splenomegalia nel 10-12% dei bambini, soprattutto quelli piccoli. In questo caso la diagnosi differenziale si pone con la leucemia, la mononucleosi infettiva, il lupus eritematoso sistemico, la sindrome dell'ipersplenismo. Nella porpora trombocitopenica idiopatica non dovrebbe esserci ingrossamento dei linfonodi, a meno che non sia associata ad una precedente infezione virale.
Porpora trombocitopenica secondaria
Come affermato in precedenza, la trombocitopenia può essere idiopatica o secondaria a una serie di cause note. La trombocitopenia secondaria, a sua volta, può essere divisa in base al numero di megacariociti.
Carenza di trombopoietina
Una rara causa congenita di trombocitopenia cronica con comparsa di numerosi megacariociti immaturi nel midollo osseo è la carenza di trombopoietina.
Il trattamento consiste in trasfusioni di plasma da donatori sani o pazienti con porpora trombocitopenica idiopatica, che determina un aumento della conta piastrinica e segni di maturazione dei megacariociti, o nella sostituzione della trombopoietina.
Diagnosi di laboratorio della porpora trombocitopenica
L'esame di laboratorio rivela una trombocitopenia inferiore a 100.000/mm3, un aumento del volume piastrinico medio (MPV) secondo un analizzatore del sangue automatico a 8,9±1,5 μm3.
Nel sangue periferico dei pazienti con porpora trombocitopenica idiopatica, oltre alla trombocitopenia, può essere presente una moderata eosinofilia. Con grave perdita di sangue, si sviluppa l'anemia.
Nella puntura del midollo osseo, che viene effettuata per escludere altre malattie oncoematologiche, si riscontrano irritazione della linea dei megacariociti e debole "allacciatura" delle piastrine con normali linee eritroidi e mieloidi. Alcuni pazienti presentano una moderata eosinofilia.
Quando si studia il profilo della coagulazione, che è facoltativo nella porpora trombocitopenica idiopatica standard, vengono rilevati un aumento del tempo di sanguinamento, una diminuzione o assenza di retrazione del coagulo e un'utilizzazione compromessa della protrombina con livelli normali di fibrinogeno, tempo di protrombina e tempo di tromboplastina parziale attivata.
Gli esami di laboratorio nei pazienti con trombocitopenia includono:
emocromo completo con striscio e determinazione della conta piastrinica;
esame della puntura del midollo osseo;
esame del sangue per ANF, anti-DNA, frazioni del complemento C3, C4, anticorpi antipiastrinici, livello di glicocalicina plasmatica, test di Coombs;
determinazione del tempo di protrombina, tempo di tromboplastina parziale attivata, livello di fibrinogeno, prodotti di degradazione del fibrinogeno;
determinazione dell'urea, creatinina ematica, esami del fegato;
esame del sangue per infezioni opportunistiche (HIV, virus Epstein-Barr, parvovirus);
esclusione delle forme secondarie di trombocitopenia.
I criteri principali per diagnosticare la porpora trombocitopenica idiopatica sono:
assenza di segni clinici di malattie sistemiche e oncoematologiche;
trombocitopenia isolata con un numero normale di eritrociti e leucociti;
numero normale o aumentato di megacariociti nel midollo osseo con elementi eritroidi e mieloidi normali;
esclusione delle forme secondarie di trombocitopenia nell'ipersplenismo, nell'anemia emolitica microangiopatica, nella sindrome DIC, nella trombocitopenia indotta da farmaci, nel lupus eritematoso sistemico, nelle infezioni virali (virus di Epstein-Barr, HIV, parvovirus).
Poiché la patogenesi della porpora trombocitopenica idiopatica si basa sulla distruzione delle piastrine cariche di autoanticorpi da parte delle cellule del sistema reticoloistiocitario, i principi fondamentali del trattamento della porpora trombocitopenica sono:
diminuzione della produzione di autoanticorpi;
alterato legame degli autoanticorpi alle piastrine;
eliminazione della distruzione delle piastrine sensibilizzate dagli anticorpi da parte delle cellule del sistema reticoloistiocitario.
In assenza di sanguinamento dalle mucose, di lieve ecchimosi dopo contusioni e di una conta piastrinica superiore a 35.000/mm3, il trattamento solitamente non è necessario. I pazienti dovrebbero evitare gli sport di contatto. Le ragazze con il ciclo mestruale traggono beneficio dai farmaci a base di progesterone ad azione prolungata (Depo-Provera e altri) per ritardare le mestruazioni di diversi mesi al fine di prevenire un intenso sanguinamento uterino.
Glucocorticoidi
Meccanismo di azione
Inibizione della fagocitosi delle piastrine con anticorpi fissati sulla loro superficie nella milza.
Produzione di anticorpi compromessa.
Compromissione del legame degli autoanticorpi con l'antigene.
Indicazioni
Sanguinamento dalle mucose; porpora grave ed ematomi abbondanti nelle sedi delle contusioni, soprattutto sulla testa e sul collo; porpora progressiva; trombocitopenia per più di 3 settimane; trombocitopenia ricorrente; conta piastrinica inferiore a 20.000/mm3 nei pazienti primari con porpora minima.
Modalità di amministrazione
Le dosi standard di corticosteroidi orali sono 1-2 mg/kg di prednisolone al giorno o 60 mg/m2 al giorno per 21 giorni con sospensione graduale. La dose viene ridotta indipendentemente dalla conta piastrinica, la remissione viene valutata alla fine del ciclo. In assenza di remissione o di diminuzione della conta piastrinica dopo aver raggiunto i livelli normali, l’esposizione ai glucocorticoidi non viene continuata. In assenza di una risposta ematologica completa durante un ciclo standard di corticosteroidi, il prednisolone viene sospeso con un “corso intermittente” (5 mg a giorni alterni dopo la pausa). È possibile ripetere il ciclo di corticosteroidi dopo 4 settimane. L'uso a lungo termine di corticosteroidi per la porpora trombocitopenica idiopatica è indesiderabile, poiché può portare alla depressione della trombocitopoiesi.
Alte dosi di corticosteroidi orali 4-8 mg/kg al giorno per 7 giorni o 10-30 mg/kg al giorno di metilprednisolone per 3-7 giorni con rapida interruzione del farmaco. Una settimana dopo i corsi vengono ripetuti (2-3 portate).
Alte dosi di corticosteroidi parenterali 10-30 mg/kg al giorno metilprednisolone o solumedrolo 500 mg/m2 al giorno per via endovenosa per 3-7 giorni nei casi gravi per un sollievo più rapido della sindrome emorragica. Se è necessario un ulteriore trattamento, il paziente viene trasferito a dosi standard per via orale.
Per i pazienti resistenti agli steroidi con porpora trombocitopenica idiopatica è possibile la “terapia pulsata” con desametasone: 6 cicli da 0,5 mg/kg al giorno (massimo 40 mg/die) per 4 giorni ogni 28 giorni, assunti per via orale.
L'efficacia dell'assunzione di cotricosteroidi, secondo vari autori, è del 50-80%. Effetti collaterali durante il loro utilizzo: sintomi di ipercorticismo, ulcera peptica, iperglicemia, ipertensione, aumento del rischio di infezione, miopatia, ipokaliemia, psicosi da steroidi, disfunzione ovarica nelle ragazze, ritardo della crescita.
Immunoglobulina endovenosa
Meccanismo di azione:
blocco reversibile dei recettori Fc dei macrofagi;
soppressione della sintesi di autoanticorpi da parte dei linfociti B;
protezione delle piastrine e/o dei megacariociti dagli anticorpi;
modulazione dell'attività helper e soppressore dei linfociti T;
soppressione del danno tissutale complemento-dipendente;
recupero da infezioni virali persistenti grazie all'introduzione di anticorpi specifici.
Indicazioni per la porpora trombocitopenica idiopatica acuta:
se possibile, esposizione in prima linea;
trombocitopenia immunitaria sintomatica neonatale;
bambini di età inferiore a 2 anni resistenti ai corticosteroidi.
Le moderne preparazioni di immunoglobuline endovenose (IVIG) devono soddisfare i requisiti dell'OMS definiti nel 1982: almeno 1000 unità di sangue, almeno il 90% di immunoglobulina G, immunoglobulina G nativa (elevata attività del frammento Fc), normale divisione dell'immunoglobulina G in sottoclassi, metà fisiologica vita. Inoltre, le IVIG dovrebbero avere una bassa attività anticomplementare e una doppia inattivazione del virus (immunoglobulina G pura).
Regimi di somministrazione di immunoglobuline per via endovenosa
Per la porpora trombocitopenica idiopatica acuta: una dose totale di 1-2 g/kg per ciclo secondo lo schema: 400 mg/kg al giorno per 5 giorni o 1 g/kg al giorno per 1-2 giorni. I bambini sotto i 2 anni di età tollerano più facilmente un protocollo di 5 giorni per l'assunzione di farmaci di 1a e 2a generazione.
Per la porpora trombocitopenica cronica idiopatica: una dose iniziale di 1 g/kg al giorno per 1-2 giorni, quindi infusioni singole alla dose di 0,4-1 g/kg, a seconda della risposta, per mantenere un livello piastrinico sicuro (più di 30.000/mm3). È utile abbinare l'uso delle IVIG a cicli alternati di corticosteroidi.
La risposta all'esposizione nei pazienti con porpora trombocitopenica idiopatica acuta si verifica nell'80-96,5% dei casi. Rispetto ai corticosteroidi, la conta piastrinica aumenta più rapidamente durante episodi emorragici di durata comparabile. Circa il 65% dei bambini con porpora trombocitopenica idiopatica resistente ai corticosteroidi ottiene una remissione a lungo termine dopo un ciclo di IVIG.
Effetti collaterali dei farmaci IVIG:
reazioni anafilattiche (in pazienti con livelli di IgA ridotti);
mal di testa (20% dei casi);
febbre con brividi (1-3% dei casi);
anemia emolitica con test di Coombs positivo.
La letteratura scientifica ha descritto un caso di sviluppo di meningite asettica dopo l'infusione di IVIG, nonché di infezione di soggetti trattati con IVIG (Gammagard\Baxter) con il virus dell'epatite C, ma dal 1994, dopo il miglioramento della tecnologia di produzione dei farmaci, tali situazioni sono non è più avvenuto.
La somministrazione profilattica di paracetamolo (10-15 mg/kg ogni 4 ore) e difenidramina (difenidramina) (1 mg/kg ogni 6-8 ore) riduce la frequenza e la gravità della febbre con brividi e la somministrazione endovenosa di desametasone alla dose di 0,15-0,3 mg/kg aiutano ad alleviare il mal di testa durante le infusioni di IVIG.
Uso combinato di glucocorticoidi e immunoglobuline endovenose
Indicazioni:
sanguinamento dalle mucose;
estese petecchie, porpora ed ecchimosi;
sintomi e/o segni di emorragia interna, in particolare emorragia intracranica.
L'uso combinato provoca un aumento più rapido della conta piastrinica rispetto a ciascun farmaco preso singolarmente. Viene utilizzato per emorragie potenzialmente letali e in preparazione a un intervento chirurgico. In casi di emergenza, come glucocorticoide può essere utilizzato il metilprednisolone 30 mg/kg al giorno per 3 giorni o il solumedrolo alla dose di 500 mg/m2.
Immunoglobuline anti-RhD
Meccanismo di azione:
blocco dei recettori Fc dei macrofagi da parte degli eritrociti carichi di anticorpi;
soppressione della formazione di anticorpi antipiastrinici;
effetto immunomodulatore.
Le condizioni per l'uso nella porpora trombocitopenica idiopatica sono i pazienti RhD positivi non splenectomizzati.
Preparazioni immunoglobuliniche anti-RhD: “WinRho” (Winnipeg, Manitoba, Canada), “NABI” (Boca Ration, FL, USA), “Partogamma” (Biagini, Pisa, Italia), “Resogam” (Genteon Pharma, Germania) .
Modalità di amministrazione:
la dose ottimale per il ciclo è di 50 mcg/kg per ciclo sotto forma di una singola infusione endovenosa o di somministrazione intramuscolare frazionata nell'arco di 2-5 giorni;
se la concentrazione di emoglobina nel sangue del paziente è inferiore a 100 g/l, la dose del farmaco è 25-40 mcg/kg per ciclo, con emoglobina 100 g/l - 40-80-100 mcg/corso;
cicli ripetuti di immunoglobulina anti-D a intervalli di 3-8 settimane per mantenere la conta piastrinica sopra 30.000/mm3.
La conta piastrinica e il livello di emoglobina vengono monitorati nei 3-4 giorni successivi all'inizio dell'esposizione. L'assenza di una risposta ematologica al primo ciclo di immunoglobuline anti-D non costituisce una controindicazione per un secondo ciclo, poiché il 25% dei pazienti che non rispondono al trattamento ottengono una risposta ematologica quando il farmaco viene ripetuto. Tra i pazienti resistenti ai corticosteroidi, il 64% raggiunge la remissione dopo un ciclo di immunoglobuline anti-D. Un aumento significativo del numero di piastrine si nota 48 ore dopo la somministrazione del farmaco, quindi non è raccomandato l'uso in situazioni pericolose per la vita.
Reazioni avverse:
sindrome simil-influenzale (febbre, brividi, mal di testa);
un calo dei livelli di emoglobina e di ematocrito dovuto all'emolisi, confermato da un test di Coombs positivo.
Non si sono verificati casi di infezione virale con l’uso di preparazioni immunoglobuliniche anti-D. Sono improbabili reazioni allergiche acute. Sono state descritte reazioni allergiche IgE-mediate e indotte da immunocomplessi. Non sono state descritte reazioni allergiche in pazienti con deficit di IgA. L'emolisi è solitamente extravascolare. Nei pochi casi di emolisi intravascolare descritti, non si è sviluppata insufficienza renale cronica. La diminuzione media dei livelli di emoglobina è di 5-20 g/l e può essere di breve durata (1-2 settimane).
L'uso dell'immunoglobulina anti-RhD è sicuro, conveniente, economico ed efficace nel 79-90% dei pazienti con porpora trombocitopenica idiopatica cronica, più spesso nei bambini che negli adulti.
Interferone alfa
L'interferone alfa-2b può essere utilizzato nel trattamento di pazienti con porpora trombocitopenica idiopatica cronica refrattari ai corticosteroidi. La risposta ematologica si ottiene nel 72% dei pazienti, compreso il 33% dei non-responder ai corticosteroidi.
Meccanismo d'azione nella porpora trombocitopenica idiopatica: soppressione della produzione di autoanticorpi dovuta all'effetto inibitorio dell'interferone-alfa-2b sulla produzione di immunoglobuline da parte dei linfociti B.
Modalità di somministrazione: 0,5-2x106 unità, a seconda dell'età, per via sottocutanea o intramuscolare 3 volte a settimana (solitamente lunedì-mercoledì-venerdì) per 1-1,5 mesi. La risposta ematologica si osserva tra il 7° e il 39° giorno dall'inizio del trattamento. In assenza di risposta ematologica il trattamento viene interrotto; se presente, viene continuato fino a 3 mesi. Dopo il completamento del corso, il farmaco viene sospeso o prescritto in una dose di mantenimento, riducendo la frequenza di somministrazione a 1-2 volte a settimana (selezionata individualmente). Se la malattia recidiva (di solito 2-8 settimane dopo la fine dell'uso), è indicato un ciclo ripetuto, che ha la stessa efficacia. La durata del trattamento di mantenimento con interferone alfa-2b in presenza di risposta ematologica non è stata determinata.
Effetti collaterali: sindrome simil-influenzale (febbre, brividi, mal di testa, mialgia), dolore e arrossamento nel sito di iniezione, tossicità epatica, inibizione della mielopoiesi (a dosi superiori a 2x106 unità), depressione negli adolescenti.
Per ridurre la gravità degli effetti collaterali (sindrome simil-influenzale), si raccomanda la somministrazione profilattica di paracetamolo prima della prima somministrazione del farmaco.
Il danazolo è un androgeno sintetico con debole attività virilizzante ed effetti immunomodulatori (ripristino della funzione T-soppressore).
Il meccanismo d'azione del danazolo nella porpora trombocitopenica idiopatica:
modula l'espressione dei recettori Fc-gamma sui fagociti mononucleati e previene la distruzione delle piastrine cariche di anticorpi;
sopprime la produzione di autoanticorpi;
ha sinergismo con i corticosteroidi, favorisce il rilascio degli steroidi dalla connessione con le globuline e aumenta il loro accesso ai tessuti.
Modalità di amministrazione:
10-20 mg/kg al giorno per via orale (300-400 mg/m2) in 2-3 dosi per 3 mesi o più per stabilizzare l'effetto.
Effetti collaterali:
acne, irsutismo, aumento di peso, tossicità epatica.
La risposta ematologica si verifica in circa la metà dei bambini con porpora trombocitopenica idiopatica cronica, compresi quelli refrattari ai corticosteroidi. L'efficacia del trattamento aumenta dopo la splenectomia. Nella maggior parte dei casi la risposta è incompleta.
Vincristina
La vincristina viene utilizzata alla dose di 0,02 mg/kg (massimo 2 mg) per via endovenosa, settimanale, per un totale di 4 iniezioni.
Vinblastina
La vinblastina viene utilizzata alla dose di 0,1 mg/kg (massimo 10 mg) per via endovenosa, settimanale, per un totale di 4 iniezioni.
Quando la vincristina e la vinblastina sono efficaci, la conta piastrinica aumenta rapidamente, spesso a livelli normali. La maggior parte dei bambini necessita di dosi ripetute del farmaco a intervalli di 2-3 settimane per mantenere una conta piastrinica sicura. Se non si ottiene risposta al trattamento entro 4 settimane, non è indicato un ulteriore utilizzo di farmaci.
La remissione ematologica completa entro 0,5-4 anni è descritta in circa il 10% dei pazienti, una risposta transitoria nella metà.
Effetti collaterali: neuropatia periferica, leucopenia, alopecia, stitichezza, necrosi se rilasciato nel tessuto sottocutaneo.
Ciclofosfamide
La ciclofosfamide (ciclofosfamide) è usata come immunosoppressore. La risposta ematologica nei pazienti con porpora trombocitopenica idiopatica cronica durante il trattamento raggiunge il 60-80% e dura più a lungo rispetto ad altri farmaci. La risposta ematologica completa dopo il completamento del trattamento si verifica nel 20-40% dei casi. I migliori risultati si ottengono nei pazienti splenectomizzati con malattia di breve durata.
Il meccanismo d'azione è la soppressione della proliferazione dei cloni linfocitici coinvolti nella risposta immunitaria.
Modalità di somministrazione: 1-2 mc/kg al giorno per via orale. La risposta ematologica si ottiene entro 2-10 settimane dall'inizio del ciclo.
Effetti collaterali: inibizione della mielopoiesi, alopecia, tossicità epatica, cistite emorragica, leucemia (complicanza a lungo termine).
Azatioprina
Nei pazienti con malattie autoimmuni, l'azatioprina viene utilizzata come immunosoppressore. Un aumento della conta piastrinica è stato osservato nel 50% dei pazienti con porpora trombocitopenica idiopatica e una risposta ematologica completa nel 10-20%.
Modalità di somministrazione: 1-5 mg/kg al giorno (200-400 mg). Fino al raggiungimento della risposta massima, la durata del trattamento può essere di 3-6 mesi. Poiché la malattia recidiva dopo la sospensione dell'uso del farmaco, è necessario un trattamento di mantenimento.
Effetti collaterali: anoressia, nausea, vomito, neutropenia moderata, linfoma (complicanza a lungo termine).
Il vantaggio di questo farmaco nei bambini è la minore incidenza di sviluppo del tumore rispetto alla ciclofosfamide (ciclofosfamide).
Ciclosporina
La ciclosporina (ciclosporina A) è un immunosoppressore non steroideo che provoca l'inibizione dell'immunità cellulare. Il farmaco agisce sui linfociti T-effettori attivati, sopprimendo la produzione di citochine (interleuchina-2, interferone gamma, fattore di necrosi tumorale).
Modalità di somministrazione: assunto per via orale alla dose di 5 mg/kg al giorno per diversi mesi. Una risposta ematologica si osserva 2-4 settimane dall'inizio del trattamento sotto forma di una certa stabilizzazione dei parametri clinici ed ematologici, una diminuzione del livello di anticorpi antipiastrinici. Le ricadute della malattia si verificano immediatamente dopo la sospensione del farmaco.
Effetti collaterali: ipomagnesiemia, ipertensione, tossicità epatica e renale, tumori secondari (complicanze a lungo termine). I gravi effetti collaterali e l'effetto inconcludente causati dall'uso della ciclosporina ne rendono indesiderabile l'uso nella porpora trombocitopenica idiopatica.
Trasfusioni di piastrine
La trasfusione di piastrine è indicata in caso di sviluppo di sintomi neurologici che indicano la possibilità di emorragia intracranica, nonché durante interventi chirurgici in pazienti con trombocitopenia profonda, resistenti al trattamento conservativo. Sebbene la durata della vita delle piastrine sia breve, le trasfusioni di piastrine possono avere un effetto emostatico temporaneo. Allo stesso tempo, il timore di aumentare la durata della porpora trombocitopenica idiopatica a causa del rischio di sensibilizzazione è solo teorico. Le trasfusioni di piastrine sono utilizzate in pazienti con porpora trombocitopenica idiopatica ad alto rischio con effetto clinico positivo. La trasfusione del concentrato piastrinico viene effettuata in dosi frazionate di 1-2 dosi all'ora o 6-8 dosi ogni 4-6 ore fino al raggiungimento della risposta clinica ed ematologica. L'effetto della trasfusione è potenziato dalla somministrazione preliminare di IVIG.
Splenectomia
In assenza di effetti dal trattamento conservativo della porpora trombocitopenica, in presenza di trombocitopenia profonda, sindrome emorragica e minaccia di sanguinamento potenzialmente letale, si consiglia ai pazienti di sottoporsi a splenectomia. La questione dell'intervento chirurgico viene decisa individualmente in ciascun caso.
Indicazioni per la splenectomia:
porpora trombocitopenica idiopatica acuta grave con presenza di sanguinamento potenzialmente letale in assenza di risposta ai farmaci;
durata della malattia superiore a 12 mesi, trombocitopenia inferiore a 10.000/mm3 e storia di sanguinamento;
porpora trombocitopenica cronica idiopatica con segni di sanguinamento e livello piastrinico costante inferiore a 30.000/mm3 senza risposta al trattamento per diversi anni.
Nei pazienti che conducono uno stile di vita attivo e sono spesso feriti, la splenectomia può essere eseguita prima.
A causa del rischio di sviluppare infezioni generalizzate dopo l’intervento chirurgico, la splenectomia viene eseguita solo se vi sono chiare indicazioni. La chirurgia è raramente necessaria entro 2 anni dalla diagnosi poiché la trombocitopenia è ben tollerata e facilmente controllata con corticosteroidi e IVIG. Il recupero spontaneo della conta piastrinica può verificarsi dopo 4-5 anni, pertanto è necessario un approccio molto attento all'operazione. Nei bambini affetti da porpora trombocitopenica cronica idiopatica si osservano casi di remissione spontanea nel 10-30% dei casi diversi mesi o anni dopo la diagnosi; negli adulti è molto rara.
La preparazione alla splenectomia comprende la somministrazione di corticosteroidi, IVIG o immunoglobuline anti-D. I corticosteroidi vengono prescritti a dose intera il giorno prima, il giorno dell'intervento e per diversi giorni successivi, poiché la maggior parte dei pazienti presenta insufficienza surrenalica dovuta al precedente utilizzo. Se si verifica un sanguinamento attivo immediatamente prima dell'intervento chirurgico, può essere necessaria una trasfusione di piastrine e globuli rossi, nonché la somministrazione di metilprednisolone (solumedrolo) alla dose di 500 mg/m2 al giorno. Prima di un'operazione pianificata, è obbligatorio un esame ecografico degli organi addominali per identificare ulteriori milze (15% dei casi) e, in casi controversi, la scansione dei radioisotopi.
Il recupero completo e duraturo della conta piastrinica dopo la splenectomia si verifica in circa il 50% dei pazienti. Un buon segno prognostico è la risposta all'assunzione di corticosteroidi e IVIG prima dell'intervento chirurgico (l'efficacia della splenectomia è dell'80-90%), così come l'assenza di anticorpi antipiastrinici dopo l'intervento. Il 25% dei bambini sottoposti a splenectomia non ottiene una risposta clinica ed ematologica e necessita di ulteriore trattamento.
È preferibile eseguire l'intervento con il metodo laparoscopico (possibile nel 90% dei pazienti) per ridurre il volume dell'intervento chirurgico, il livello di perdita di sangue chirurgica, fornire al paziente un ritorno più rapido alla vita attiva e abbreviare la durata del ricovero ospedaliero. . La cicatrice postoperatoria è lunga circa 1 cm e non provoca disagio.
I casi di morte per infezioni batteriche nel tardo periodo postoperatorio, soprattutto nei bambini sottoposti a splenectomia prima dei 5 anni di età, ammontano a 1:300 pazienti all'anno. La maggior parte di essi si verifica entro 2 anni dall’intervento. Le cause principali comprendono le infezioni da pneumococco e meningococco, che si sviluppano come sepsi fulminante con coagulazione intravascolare disseminata ed emorragie nelle ghiandole surrenali. Pertanto, non più tardi di due settimane prima dell'intervento chirurgico, si raccomanda la somministrazione di vaccini pneumococcici, meningococcici e Haemophilus influenzae e l'uso profilattico a lungo termine, almeno 2 anni, della benzilpenicillina dopo la splenectomia. Alcuni autori suggeriscono di limitare la somministrazione di bicillina-5 (benzatina benzilpenicillina + benzilpenicillina procaina) mensilmente per 6 mesi dopo l'intervento.
Una possibile alternativa alla splenectomia è l'occlusione endovascolare della milza, che può essere eseguita anche in pazienti con trombocitopenia grave. Per ottenere un effetto clinico ed ematologico duraturo, è necessario eliminare gradualmente il 90-95% del parenchima dell'organo. La reattività immunologica del corpo dopo l'occlusione endovascolare della milza è preservata grazie al funzionamento del 2-5% del tessuto splenico, che mantiene l'afflusso di sangue grazie ai collaterali, che è importante nella pratica pediatrica. È possibile utilizzare l'occlusione endovascolare prossimale della milza diversi giorni prima della splenectomia per ridurre il rischio di un intervento chirurgico.
Plasmaferesi
Nei pazienti con trombocitopenia persistente ed emorragia pericolosa per la vita nonostante l'intervento medico e la splenectomia, la reinfusione del plasma attraverso colonne di proteina A può essere utilizzata per rimuovere rapidamente gli anticorpi antipiastrinici. Nei pazienti con porpora trombocitopenica idiopatica grave, ciò accelera l’eliminazione del fattore antipiastrinico circolante.
Trattamento dei bambini con emorragia pericolosa per la vita:
trasfusioni di piastrine;
solyumedrol 500 mg/m2 al giorno per via endovenosa in 3 iniezioni;
immunoglobulina endovenosa 2 g/kg per ciclo;
splenectomia immediata.
Queste misure possono essere eseguite singolarmente o in combinazione a seconda della gravità e della risposta al trattamento.
Prognosi nei bambini con porpora trombocitopenica idiopatica
Nel 70-80% dei pazienti, la remissione avviene entro 6 mesi, nel 50% entro 1 mese dall'esordio della malattia.
L'inizio della remissione spontanea dopo un anno di malattia è insolito, ma può essere notato anche dopo diversi anni.
La prognosi della malattia non dipende dal sesso, dalla gravità della condizione iniziale e dal rilevamento dell'eosinofilia nel midollo osseo.
Una volta identificata la causa della porpora trombocitopenica idiopatica, la prognosi dipende dalla sua eliminazione.
Circa il 50-60% dei pazienti con porpora trombocitopenica idiopatica cronica si stabilizzerà senza alcun trattamento o splenectomia
Un tipo di diatesi emorragica, caratterizzata da una carenza di piastrine del sangue rosso - piastrine, spesso causata da meccanismi immunitari. I segni della porpora trombocitopenica sono emorragie spontanee, multiple, polimorfiche nella pelle e nelle mucose, nonché sanguinamenti nasali, gengivali, uterini e di altro tipo. Se si sospetta porpora trombocitopenica, vengono valutati i dati anamnestici e clinici, gli indicatori generali dell'emocromo, i coagulogrammi, l'ELISA, la microscopia degli strisci di sangue e la puntura del midollo osseo. Per scopi terapeutici, ai pazienti vengono prescritti corticosteroidi, farmaci emostatici, terapia citostatica e viene eseguita la splenectomia.

informazioni generali
La porpora trombocitopenica (malattia di Werlhof, trombocitopenia benigna) è una patologia ematologica caratterizzata da una carenza quantitativa di piastrine nel sangue, accompagnata da una tendenza al sanguinamento e dallo sviluppo della sindrome emorragica. Con la porpora trombocitopenica, il livello delle piastrine nel sangue periferico scende significativamente al di sotto del livello fisiologico - 150x10 9 / l con un numero normale o leggermente aumentato di megacariociti nel midollo osseo. In termini di frequenza, la porpora trombocitopenica è al primo posto tra le altre diatesi emorragiche. La malattia si manifesta solitamente nell'infanzia (con un picco nel periodo precoce e prescolare). Negli adolescenti e negli adulti, la patologia viene rilevata 2-3 volte più spesso tra le donne.
La classificazione della porpora trombocitopenica tiene conto delle sue caratteristiche eziologiche, patogenetiche e cliniche. Esistono diverse varianti: porpora trombocitopenica idiopatica (malattia di Werlhof), porpora trombocitopenica iso, trans, etero e autoimmune, complesso dei sintomi di Werlhof (trombocitopenia sintomatica).
Secondo il corso si distinguono le forme acute, croniche e ricorrenti. La forma acuta è più tipica dell'infanzia, dura fino a 6 mesi con normalizzazione dei livelli piastrinici nel sangue e non presenta ricadute. La forma cronica dura più di 6 mesi ed è più comune nei pazienti adulti; ricorrente - ha un decorso ciclico con ripetizioni di episodi di trombocitopenia dopo la normalizzazione dei livelli piastrinici.
Cause della porpora trombocitopenica
Nel 45% dei casi si verifica porpora trombocitopenica idiopatica, che si sviluppa spontaneamente, senza una ragione apparente. Nel 40% dei casi la trombocitopenia è preceduta da diverse malattie infettive (virali o batteriche), subite circa 2-3 settimane prima. Nella maggior parte dei casi si tratta di infezioni del tratto respiratorio superiore di origine non specifica, nel 20% specifiche (varicella, morbillo, rosolia, parotite, mononucleosi infettiva, pertosse). La porpora trombocitopenica può complicare il decorso della malaria, della febbre tifoide, della leishmaniosi e dell'endocardite settica. A volte la porpora trombocitopenica appare sullo sfondo dell'immunizzazione - attiva (vaccinazione) o passiva (somministrazione di γ - globulina). La porpora trombocitopenica può essere scatenata dall'assunzione di farmaci (barbiturici, estrogeni, arsenico, mercurio), esposizione prolungata ai raggi X (isotopi radioattivi), interventi chirurgici estesi, traumi ed eccessiva insolazione. Sono stati segnalati casi familiari della malattia.
La maggior parte delle varianti della porpora trombocitopenica sono di natura immunitaria e sono associate alla produzione di anticorpi antipiastrinici (IgG). La formazione di complessi immunitari sulla superficie delle piastrine porta alla rapida distruzione delle piastrine del sangue, riducendone la durata di vita a diverse ore invece dei 7-10 giorni normalmente.
La forma isoimmune della porpora trombocitopenica può essere causata dall'ingresso di piastrine “estranee” nel sangue durante trasfusioni ripetute di sangue o piastrine, nonché dall'incompatibilità antigenica delle piastrine della madre e del feto. La forma eteroimmune si sviluppa quando la struttura antigenica delle piastrine viene danneggiata da vari agenti (virus, farmaci). La variante autoimmune della porpora trombocitopenica è causata dalla comparsa di anticorpi contro i propri antigeni piastrinici invariati e di solito è associata ad altre malattie della stessa origine (LES, anemia emolitica autoimmune). Lo sviluppo della trombocitopenia transimmune nei neonati è provocato dal passaggio di autoanticorpi antipiastrinici attraverso la placenta di una madre affetta da porpora trombocitopenica.
La carenza piastrinica nella porpora trombocitopenica può essere associata a danno funzionale ai megacariociti e ad un'interruzione nel processo di eliminazione delle piastrine rosse. Ad esempio, il complesso dei sintomi di Verlhof è causato dall'inefficacia dell'ematopoiesi nell'anemia (carenza di B-12, aplastica), leucemia acuta e cronica, malattie sistemiche degli organi ematopoietici (reticolosi), metastasi del midollo osseo di tumori maligni.
Con la porpora trombocitopenica si verifica un'interruzione della formazione di tromboplastina e serotonina, una diminuzione della contrattilità e un aumento della permeabilità della parete capillare. Ciò è associato al prolungamento del tempo di sanguinamento, all’interruzione della formazione di trombi e alla retrazione del coagulo sanguigno. Durante le riacutizzazioni emorragiche, il numero delle piastrine nel farmaco diminuisce fino a formare singole cellule; durante il periodo di remissione viene ripristinato a un livello inferiore al normale.
Sintomi della porpora trombocitopenica
La porpora trombocitopenica si manifesta clinicamente quando il livello delle piastrine scende al di sotto di 50x10 9 /l, solitamente 2-3 settimane dopo l'esposizione al fattore eziologico. Il sanguinamento di tipo petecchiale (lividi) è caratteristico. Nei pazienti con porpora trombocitopenica compaiono emorragie multiple indolori sotto la pelle, nelle mucose (versione "secca"), nonché sanguinamenti (versione "umida"). Si sviluppano spontaneamente (spesso di notte) e la loro gravità non corrisponde alla forza dell'impatto traumatico.
Le eruzioni emorragiche sono polimorfiche (da petecchie ed ecchimosi minori a grandi lividi e contusioni) e policrome (dal blu violaceo brillante al giallo-verde pallido, a seconda del momento della comparsa). Molto spesso, le emorragie si verificano sulla superficie anteriore del busto e degli arti, raramente sul viso e sul collo. Le emorragie vengono rilevate anche sulla mucosa delle tonsille, sul palato molle e duro, sulla congiuntiva e sulla retina, sul timpano, sul tessuto adiposo, sugli organi parenchimali e sulle membrane sierose del cervello.
Il sanguinamento intenso è patognomonico: sanguinamento nasale e gengivale, sanguinamento dopo l'estrazione del dente e la tonsillectomia. Possono comparire emottisi, vomito con sangue, diarrea e sangue nelle urine. Nelle donne, il sanguinamento uterino di solito prevale sotto forma di menorragia e metrorragia, nonché sanguinamento ovulatorio nella cavità addominale con sintomi di gravidanza ectopica. Immediatamente prima delle mestruazioni compaiono elementi emorragici cutanei, sangue dal naso e altri sanguinamenti. La temperatura corporea rimane normale, è possibile la tachicardia. Nella porpora trombocitopenica è presente una moderata splenomegalia. Con sanguinamento abbondante si sviluppa anemia degli organi interni, iperplasia del midollo osseo rosso e megacariociti.
La forma medicinale si manifesta subito dopo l'assunzione del farmaco, dura da 1 settimana a 3 mesi con guarigione spontanea. La porpora trombocitopenica da radiazioni è caratterizzata da una grave diatesi emorragica con la transizione del midollo osseo in uno stato ipo e aplastico. La forma infantile (nei bambini sotto i 2 anni di età) ha un esordio acuto, grave, spesso cronico e trombocitopenia grave (9/l).
Durante la porpora trombocitopenica si identificano periodi di crisi emorragica, remissione clinica e clinico-ematologica. Durante una crisi emorragica, si notano sanguinamenti e alterazioni di laboratorio; durante il periodo di remissione clinica, sullo sfondo della trombocitopenia, non compaiono emorragie. Con la remissione completa, non si verificano sanguinamenti o alterazioni di laboratorio. Con la porpora trombocitopenica con grande perdita di sangue, si osserva anemia postemorragica acuta, con una forma cronica a lungo termine - anemia cronica da carenza di ferro.
La complicazione più grave, l'emorragia cerebrale, si sviluppa improvvisamente e progredisce rapidamente, accompagnata da vertigini, mal di testa, vomito, convulsioni e disturbi neurologici.
Diagnosi di porpora trombocitopenica
La diagnosi di porpora trombocitopenica viene stabilita da un ematologo tenendo conto dell'anamnesi, delle caratteristiche del decorso e dei risultati degli esami di laboratorio (analisi clinica del sangue e delle urine, coagulogramma, ELISA, microscopia di strisci di sangue, puntura del midollo osseo).
La porpora trombocitopenica è indicata da una forte diminuzione del numero di piastrine nel sangue (9/l), da un aumento del tempo di sanguinamento (>30 minuti), del tempo di protrombina e dell'aPTT, da una diminuzione del grado o dell'assenza di retrazione del coagulo. La conta dei globuli bianchi è solitamente entro limiti normali; l'anemia si manifesta con una significativa perdita di sangue. Al culmine della crisi emorragica vengono rilevati test endoteliali positivi (pizzico, laccio emostatico, puntura). Uno striscio di sangue rivela un aumento delle dimensioni e una diminuzione della granularità delle piastrine. Nelle preparazioni di midollo osseo rosso si rileva un numero normale o aumentato di megacariociti, la presenza di forme immature e l'allacciamento piastrinico in pochi punti. La natura autoimmune della porpora è confermata dalla presenza di anticorpi antipiastrinici nel sangue.
La porpora trombocitopenica si differenzia dai processi aplastici o infiltrativi del midollo osseo, dalla leucemia acuta, dalle trombocitopatie, dal LES, dall'emofilia, dalla vasculite emorragica, dall'ipo- e disfibrinogenemia, dal sanguinamento uterino giovanile.
Trattamento e prognosi della porpora trombocitopenica
Per la porpora trombocitopenica con trombocitopenia isolata (piastrine >50x10 9 /l) senza sindrome emorragica, il trattamento non viene effettuato; per la trombocitopenia moderata (30-50 x10 9 /l), la terapia farmacologica è indicata in caso di aumentato rischio di sanguinamento (ipertensione arteriosa, ulcere gastriche e duodenali). Se il livello delle piastrine è 9/L, il trattamento viene effettuato senza ulteriori indicazioni in ambito ospedaliero.
Il sanguinamento viene fermato mediante la somministrazione di farmaci emostatici e l'applicazione locale di una spugna emostatica. Per frenare le reazioni immunitarie e ridurre la permeabilità vascolare, i corticosteroidi vengono prescritti in dose decrescente; globuline iperimmune. In caso di grandi perdite di sangue sono possibili trasfusioni di plasma e globuli rossi lavati. Le infusioni di piastrine non sono indicate per la porpora trombocitopenica.
Nei pazienti con forma cronica con recidive di forti emorragie ed emorragie negli organi vitali, viene eseguita la splenectomia. È possibile prescrivere immunosoppressori (citostatici). Il trattamento della porpora trombocitopenica, se necessario, deve essere combinato con la terapia per la malattia di base.
Nella maggior parte dei casi, la prognosi della porpora trombocitopenica è molto favorevole, il recupero completo è possibile nel 75% dei casi (nei bambini - 90%). Le complicazioni (ad esempio l'ictus emorragico) si verificano nella fase acuta, creando un rischio di morte. La porpora trombocitopenica richiede un monitoraggio costante da parte di un ematologo, i farmaci che influenzano le proprietà di aggregazione delle piastrine (acido acetilsalicilico, caffeina, barbiturici), sono esclusi gli allergeni alimentari, si presta cautela quando si vaccinano i bambini e l'insolazione è limitata.
RCHR (Centro Repubblicano per lo Sviluppo Sanitario del Ministero della Salute della Repubblica del Kazakistan)
Versione: Protocolli clinici del Ministero della Salute della Repubblica del Kazakistan - 2016
Porpora trombocitopenica idiopatica (D69.3)
Oncologia infantile, Pediatria
informazioni generali
Breve descrizione
Approvato
Commissione mista sulla qualità sanitaria
Ministero della Sanità e dello Sviluppo Sociale della Repubblica del Kazakistan
del 29 novembre 2016
Protocollo n. 16
Trombocitopenia immune- una malattia autoimmune caratterizzata da trombocitopenia isolata (meno di 100.000/μl) con un numero costante/aumentato di megacariociti nel midollo osseo e la presenza di anticorpi antipiastrinici sulla superficie delle piastrine e nel plasma dei pazienti, che di solito agiscono sulla membrana complessi glicoproteici IIb/IIIa e/o GPIb/ IX, che porta alla distruzione delle piastrine da parte delle cellule del sistema cellulare mononucleare fagocitico, manifestata dalla sindrome emorragica.
Correlazione dei codici ICD-10 e ICD-9
| ICD-10 | ICD-9 | ||
| Codice | Nome | Codice | Nome |
| D69.3 | trombocitopenia immunitaria | - | - |
Data di sviluppo del protocollo: 2016
Utenti del protocollo: Medici di base, terapisti, cardiologi, ematologi, pediatri, oncologi.
Scala del livello di evidenza
| UN | Una meta-analisi di alta qualità, revisione sistematica di RCT o ampi RCT con una probabilità molto bassa (++) di bias, i cui risultati possono essere generalizzati a una popolazione appropriata. |
| IN | Revisione sistematica di alta qualità (++) di studi di coorte o caso-controllo, o studi di coorte o caso-controllo di alta qualità (++) con rischio molto basso di bias, o RCT con basso (+) rischio di bias, il i cui risultati possono essere generalizzati ad una popolazione appropriata. |
| CON |
Studio di coorte o caso-controllo o studio controllato senza randomizzazione con basso rischio di bias (+). I cui risultati possono essere generalizzati alla popolazione rilevante o a RCT con rischio di bias molto basso o basso (++ o +), i cui risultati non possono essere generalizzati direttamente alla popolazione rilevante. |
| D | Serie di casi o studi non controllati o opinioni di esperti. |
XI Congresso KARM-2019: Trattamento dell'infertilità. VRT
Classificazione
ClassificazioneSocietà americana di ematologia, 2013:
Con il flusso:
· nuova identificazione - durata fino a 3 mesi;
· ITP persistente (protratto) - durata 3-12 mesi;
ITP cronica - durata superiore a 12 mesi.
Secondo la gravità della sindrome emorragica:
· grave - pazienti con sanguinamento clinicamente significativo, indipendentemente dal livello piastrinico. Casi accompagnati da sintomi di sanguinamento all'esordio della malattia, che richiedono l'inizio della terapia, oppure casi di ripresa del sanguinamento con necessità di ulteriori benefici terapeutici con vari farmaci che aumentano il numero delle piastrine, o un aumento del dosaggio del farmaco farmaci utilizzati.
· refrattario - incapacità di ottenere una risposta o una risposta completa (piastrine inferiori a 30x109/l) alla terapia dopo splenectomia; perdita di risposta dopo splenectomia e necessità di trattamento medico per ridurre al minimo il sanguinamento clinicamente significativo. In questo caso è necessario ripetere l'esame per escludere altre cause di trombocitopenia e confermare la diagnosi di ITP. Si riscontra principalmente negli adulti.
Di fasi; Standardizzazione dell'ITP, settembre 2006 IMBACH]:

Diagnostica (ambulatorio)
DIAGNOSTICA AMBULATORIALE
Criteri diagnostici: ATTENZIONE! La trombocitopenia immunitaria primaria viene diagnosticata quando le piastrine diminuiscono a meno di 100x109/l, con l'esclusione di altre cause di trombocitopenia.
Criteri diagnostici per la diagnosi:
Denunce, contestazioni:
aumento del sanguinamento dalle mucose;
Anamnesi:
· sangue dal naso, sanguinamento delle gengive;
· menorragia, metrorragia;
· emorragie nella sclera;
· emorragie cerebrali;
· ematuria;
· sanguinamento dal tratto gastrointestinale (vomito sanguinolento, melena);
· eruzioni emorragiche sotto forma di petecchie ed ecchimosi sulla pelle.
Esame fisico:
Ispezione generale:
Caratteristiche della sindrome emorragica cutanea:
· localizzazione e dimensione delle petecchie e delle contusioni;
· presenza di emorragie sulla mucosa orale, congiuntive;
· sangue che scorre lungo la parte posteriore della gola;
· anomalie della struttura del viso (viso triangolare, occhi piccoli, epicanto, piccoli tratti somatici) e degli arti (anomalie del 1° dito, sei dita, sindattilia, clinodattilia);
Ricerca di laboratorio:
· CBC con calcolo manuale della formula leucocitaria e della morfologia piastrinica - nell'emogramma si nota trombocitopenia isolata: una diminuzione delle piastrine inferiore a 100x10 9 / l senza cambiamenti nei parametri dei leucociti e dell'eritrogramma. In alcuni casi si possono registrare anemia postemorragica e alterazioni del leucogramma associate a una concomitante malattia infettiva o allergia;
NO.
Algoritmo diagnostico a livello ambulatoriale:
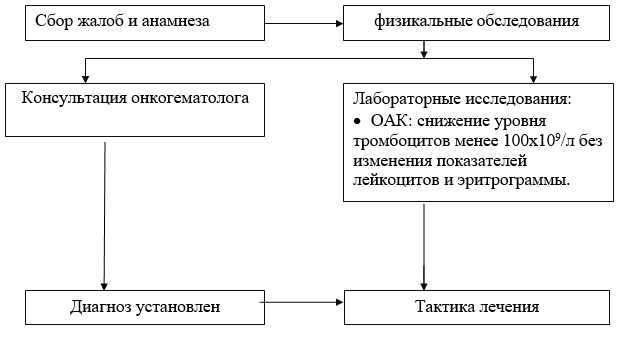
Diagnostica (ospedale)
DIAGNOSTICA A LIVELLO DEGENZIALE
Criteri diagnostici:
Denunce, contestazioni: vedere il livello ambulatoriale.
Anamnesi:
Durata e natura del sanguinamento;
· vaccinazione (in particolare vaccinazione combinata contro morbillo, parotite e rosolia) 2-3 settimane prima dello sviluppo della sindrome emorragica;
· trasferito (virale respiratorio, rosolia, mononucleosi infettiva) 2-3 settimane prima dello sviluppo della sindrome emorragica;
· uso di farmaci (in particolare eparina) nelle ultime 2-3 settimane;
presenza di dolore osseo e perdita di peso;
Esame fisico: vedere il livello ambulatoriale .
Ricerca di laboratorio:
· UAC con calcolo manuale della formula leucocitaria e della morfologia piastrinica - l'emogramma evidenzia trombocitopenia isolata - una diminuzione delle piastrine inferiore a 100x109/l senza modificare i valori leucocitari ed eritrografici. In alcuni casi si possono registrare anemia postemorragica e alterazioni del leucogramma associate a una concomitante malattia infettiva o allergia;
Studi strumentali: NO.
Algoritmo diagnostico a livello stazionario: NO.
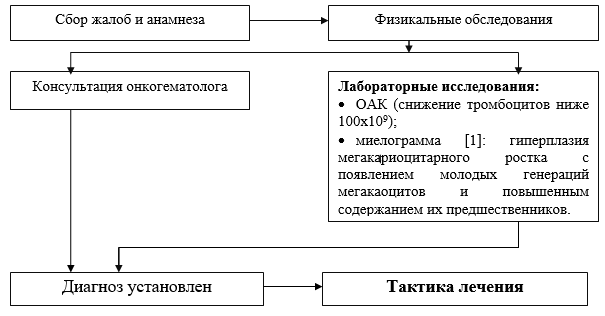
Elenco delle misure diagnostiche di base eseguite a livello ospedaliero:
· Emocromo (conta delle piastrine e dei reticolociti in uno striscio);
· gruppo sanguigno e fattore Rh;
· esame del sangue biochimico (proteine, albumina, ALT, ACaT, bilirubina, creatinina, urea, destrosio);
· mielogramma: iperplasia della linea dei megacariociti con comparsa di giovani generazioni di megacaociti e aumento del contenuto dei loro precursori;
· durata del sanguinamento secondo Sukharev;
·OAM;
· ELISA per marcatori di epatite virale (HbsAg);
· ELISA per marcatori dell'epatite virale HCV;
· ELISA per i marcatori dell'HIV.
Elenco degli ulteriori esami diagnostici eseguiti a livello ospedaliero:
· analisi biochimiche: GGTP, elettroliti;
· coagulogramma;
· ELISA per anticorpi antitrombotici;
Immunofenotipizzazione delle cellule del sangue periferico;
· immunogramma;
· anticorpi antifosfolipidi;
· PCR per le infezioni virali (epatite virale, citomegalovirus, virus dell'herpes simplex, virus Epstein-Barr, virus Varicella/Zoster);
· ecocardiografia;
· Ultrasuoni degli organi addominali (fegato, milza, pancreas, cistifellea, linfonodi, reni), mediastino, retroperitoneo e pelvi - per escludere emorragia negli organi interni;
· tomografia computerizzata del cervello: eseguita se vi è il sospetto di emorragia intracranica - mal di testa, vomito, paresi, disturbi della coscienza; escludere l'ictus;
· Ultrasuoni della OBP.
Diagnosi differenziale
| Diagnosi | Motivazione della diagnosi differenziale | Sondaggio | Criteri di esclusione della diagnosi |
| Sindrome TAR | Caratterizzato dalla patologia dei megacariociti e delle piastrine con la loro ipoplasia e disfunzione, che porta al sanguinamento | Raccolta dei reclami e dell'anamnesi, metodo dell'esame fisico. | Caratterizzato dall'assenza di ossa radiali, patologia congenita dei megacariociti e delle piastrine con la loro ipoplasia e disfunzione, che porta al sanguinamento. La malattia dei bambini è spesso accompagnata da anomalie congenite degli organi (spesso difetti cardiaci) |
| Anemia aplastica | Negli strisci di sangue, la trombocitopenia isolata è spesso profonda finché non vengono rilevate singole piastrine. | CBC con conteggio della formula leucocitaria, reticolociti. Mielogramma, biopsia con trapano. | L'aspirato di midollo osseo è povero di elementi nucleari. La percentuale totale di elementi cellulari è ridotta. Nelle preparazioni istologiche di campioni bioptici trapanati delle ossa iliache, l'aplasia del midollo osseo con sostituzione del tessuto adiposo esclude la ITP. I livelli di ferro sono normali o elevati. |
| Sindrome mielodisplasica | Sindrome emorragica | Emocromo (con conta leucocitaria, conta reticolocitaria), mielogramma, biopsia trivellata. | La MDS è caratterizzata da segni di dispoiesi, eccesso di blasti nel midollo osseo, aberrazioni cromosomiche, che escludono la ITP |
| Ematoblastosi | Pancitopenia, sindrome emorragica | CBC (con conta leucocitaria, conta dei reticolociti). Mielogramma. | I risultati della citometria a flusso, dell'esame immunoistochimico e istologico del midollo osseo escludono la ITP. |
| Emoglobinuria parossistica notturna | Sindrome emorragica |
UAC; Chimica del sangue; Coagulogramma; OAM; IFT su PNG. |
La EPN è caratterizzata da emosiderinuria, emoglobinuria, aumento dei livelli di bilirubina, LDH e diminuzione o assenza di aptoglobina. Raramente si osserva sanguinamento; è tipica l'ipercoagulazione (attivazione degli induttori di aggregazione). Escluso se non è presente alcun clone EPN in base ai risultati IFT. |
| Anemia megaloblastica. | trombocitopenia |
Emocromo + morfologia del sangue periferico; Mielogramma; Esame del sangue biochimico (livelli di cianocobalamina e acido folico). |
I segni indiretti caratteristici dell'anemia megaloblastica sono un aumento del contenuto medio di emoglobina negli eritrociti, un aumento del volume medio degli eritrociti e il tipo megaloblastico dell'ematopoiesi secondo il mielogramma. A differenza della PTI, nell'anemia megaloblastica, nonostante la trombocitopenia, non è presente la sindrome emorragica. |
| Porpora trombotica trombocitopenica. | Sindrome emorragica |
UAC; Ecografia della OBP; Valutazione dello stato neurologico; Radiografia delle articolazioni. |
Viene esclusa sulla base di sintomi neurologici, formazione di coaguli di sangue multipli, sindrome articolare e spesso ingrossamento del fegato e della milza. |
Cure all'estero
Ottieni cure in Corea, Israele, Germania, Stati Uniti
Cure all'estero
Ottieni consigli sul turismo medico
Trattamento
Farmaci (principi attivi) utilizzati nel trattamento
| Spugna emostatica |
| Azitromicina |
| Alemtuzumab |
| Amoxicillina |
| Aciclovir |
| Desametasone |
| Immunoglobulina G umana normale (Immunoglobulina G umana normale) |
| Captopril |
| Acido clavulanico |
| Kolekaltsiferolo |
| Concentrato piastrinico (CT) |
| Acido micofenolico (micofenolato mofetile) |
| Omeprazolo |
| Pancreatina |
| Paracetamolo |
| Piperacillina |
| Prednisolone |
| Rituximab |
| Tazobactam |
| Acido tranexamico |
| Trombino |
| Fluconazolo |
| Ceftazidima |
| Ciclosporina |
| Ciclofosfamide |
| Eltrombopag |
| Etamsylate |
Trattamento (ambulatorio)
TRATTAMENTO AMBULATORIO
Tattiche di trattamento: NO.
− Trattamento non farmacologico: NO.
− Trattamento farmacologico: NO.
Algoritmo di azione in situazioni di emergenza:
· consultazione con un oncoematologo - se si sospetta ematoblastosi;
· consultazione con un ginecologo - per metrorragia, menorragia;
Trattamento (ambulanza)
DIAGNOSI E TRATTAMENTO IN FASE DI EMERGENZA
Misure diagnostiche:
· raccolta dei reclami e dell'anamnesi;
· esame fisico.
Trattamento farmacologico:
· terapia sintomatica ,
secondo le linee guida IMCI - OMS per la gestione delle malattie più comuni negli ospedali di livello primario, adattate alle condizioni della Repubblica del Kazakistan.
Trattamento (ospedaliero)
TRATTAMENTO OSPEDALIERO
Tattiche di trattamento:
Per la trombocitopenia immune, le tattiche di trattamento iniziano con la prescrizione di un farmaco ormonale (prednisolone). Con una risposta favorevole al trattamento, la conta piastrinica aumenta (di solito nei giorni 7-10) e rimane ad un livello elevato anche dopo la sospensione del farmaco. Se la remissione non si verifica, viene prescritta l'immunoterapia: immunoglobulina per via endovenosa. Se non è possibile portare il paziente in remissione con la terapia farmacologica entro 6 mesi, si consiglia la splenectomia. Nei casi più gravi della malattia, la splenectomia può essere eseguita in una data precedente.
Per prendere una decisione sulla tattica terapeutica, un gruppo internazionale di esperti ha sviluppato una scala di sanguinamento e raccomandazioni per l'approccio
alla terapia:
| Sanguinamento/qualità della vita | Approccio terapeutico |
|
Grado 1. Sanguinamento minore<100 петехий и/или < 5 мелких синяков (<3 см в диаметре); отсутствие кровоточивости слизистых |
Osservazione |
|
Grado 2. Leggero sanguinamento. Petecchie multiple > 100; e/o >5 grandi lividi (>3 cm di diametro); nessun sanguinamento delle mucose |
Osservazione o, in alcuni pazienti, terapia di stabilizzazione della membrana |
|
Grado 3. Sanguinamento moderato. La presenza di sanguinamento delle mucose, uno stile di vita “pericoloso”. |
Consultazione con un ematologo |
|
Grado 4. Sanguinamento delle mucose o sospetto sanguinamento interno |
Trattamento di tutti i pazienti in ambito ospedaliero |
Trattamento non farmacologico:
Modalità: II.III;
Dieta: № 11.
Trattamento farmacologico
Trattamento a seconda della gravità:
Utilizzare la dose standard di prednisolone per un massimo di 14 giorni/dose aumentata per 4 giorni
Trattamento di prima linea per la ITP:
| Droghe | Dose | Durata della terapia |
UD, collegamento |
| Prednisolone | 0,25mg/kg | 21 giorni | Grado A |
| 2mg/kg | 14 giorni con ritiro graduale | ||
| 60mg/m2 | 21 giorni | ||
| 4mg/kg | 7 giorni con ritiro graduale | ||
| 4mg/kg | 4 giorni | ||
| Metilprednisolone | 30 o 50 mg/kg | 7 giorni | Grado A |
| 20-30 mg/kg | 2 - 7 giorni | ||
| 30 mg/kg | 3 giorni | ||
| IVIG | 0,8-1 g/kg | 1-2 giorni | Grado A |
| 0,25 g/kg | Una volta | ||
| 0,4 g/kg | 5 giorni | ||
| Anti-D | 25 µg/kg | 2 giorni | Grado A |
| 50-60 mcg/kg | Una volta | ||
| 75 mcg/kg | Una volta | ||
| Desametasone | 20 - 40 mg/kg/giorno | per 4 giorni consecutivi (ogni mese, 6 cicli) | Grado A |
PTI persistente e cronica:
· regimi terapeutici con glucocorticoidi: alte dosi di metilprednisolone IV 30 mg/kg x 3 giorni, poi 20 mg/kg x 4 giorni;
· IVIT può essere utilizzato anche per CITP, prima di interventi chirurgici, estrazioni dentarie/in caso di lesioni. I regimi per l'utilizzo dell'IVIT per la cITP sono identici a quelli per la ITP di nuova insorgenza;
· la dose consigliata di IVIT è di 0,8-1,0 g/kg di peso corporeo, seguita da somministrazioni ripetute entro 48 ore, se dopo la prima somministrazione il livello piastrinico non è superiore a 20 x 109/l.
Terapia farmacologica di seconda linea:
Rituximab(UD-B):
· dose singola: 375 mg/m2/settimana, durata del ciclo: 4 settimane (4 iniezioni in totale);
Indicazioni:
· non-responder ad alte dosi di desametasone;
· se vi sono controindicazioni alla splenectomia;
· decorso ricorrente e refrattario della ITP.
Ciclosporina A:
· 2,5 - 3 mg/kg/giorno. In combinazione con Prednisolone (UD-B)
Ciclofosfamide: 200 mg/m2 1 volta al giorno;
Indicazioni:
· in pazienti resistenti alla terapia ormonale e/o dopo splenectomia;
· ITP secondario.
Micofenolato Mofetina: 20-40 mg/kg, durata del corso 30 giorni.
Indicazioni:
· alcuni pazienti con scopi antiproliferativi e immunosoppressivi.
Terapia farmacologica di terza linea:
Agonisti dei recettori TPO(UD-A):
· Eltrombopag 25-75 mg per via orale 1-10 mg/kg/settimana.
Alemtuzumab*:
· terapia alternativa per la cITP e la ITP refrattaria.ATTENZIONE! utilizzato sullo sfondo della terapia di accompagnamento (antibatterica, antifungina, antivirale).
Elenco dei medicinali essenziali:
| INN del farmaco | Modulo per il rilascio |
UD, collegamento |
| Farmaci immunosoppressori | ||
| desametasone |
compresse da 0,5 mg soluzione 4 mg/2 ml |
UD B |
| prednisolone | Compresse da 5 mg | UD A |
| per somministrazione endovenosa 10% 2 g/20 ml | UD A | |
| immunoglobuline umane Ig G | per somministrazione endovenosa 10% 5 g/50 ml | UD A |
| ciclofosfamide | polvere per la preparazione di soluzione per somministrazione endovenosa 500 mg | UD S |
| micofenolato mofetile | capsule da 250 e 500 mg | UD S |
| rituximab |
flaconi da 10 ml/100 mg flaconi da 50 ml/500 mg |
UD B |
| ciclosporina A | capsule da 25 mg, 50 mg, 100 mg | UD B |
| Eltrombopag | compresse da 31,9 mg e 63,8 mg | UD A |
| Alemtuzimab (dopo la registrazione nella Repubblica del Kazakistan) | soluzione per infusione 1ml | UD A |
| Farmaci antifungini(secondo indicazioni) | ||
| fluconazolo | soluzione per iniezione endovenosa, 50 ml, 2 mg/ml, capsule 150 mg | UD B |
| Antimicrobici utilizzato per prevenire lo sviluppo di complicanze purulento-settiche, nonché dopo aver determinato la sensibilità agli antibiotici | ||
|
azitromicina O |
compressa/capsula, 500 mg, polvere liofilizzata per la preparazione di soluzione per infusione endovenosa, 500 mg; | UD B |
|
piperacillina/tazobactam O |
polvere per la preparazione della soluzione iniettabile per somministrazione endovenosa 4,5 g | UD B |
|
ceftazidima O |
polvere per la preparazione della soluzione iniettabile per somministrazione endovenosa 1000 mg | UD B |
| amoxocillina + acido clavulanico |
compressa rivestita con film, 500 mg/125 mg, polvere per sospensione orale 135 mg/5 ml, polvere per la preparazione di soluzione per somministrazione endovenosa e intramuscolare 600 mg. |
UD B |
| Antivirale ( secondo le indicazioni, in caso di infezione) | ||
| aciclovir | crema per uso esterno 5% -5,0, compressa 200 mg, polvere per soluzione per infusione 250 mg; | UD S |
| Medicinali che influenzano il sistema di coagulazione del sangue | ||
| fibrinogeno+trombina | spugna emostatica, misura 7*5*1, 8*3; | UD B |
Elenco di medicinali aggiuntivi:
| INN del farmaco |
Via di somministrazione |
UD, collegamento |
| omeprazolo (prevenzione della terapia antiulcera) | per somministrazione orale 20 mg | UD B |
| pancreatina (per la gastrite, migliora il processo di digestione con la terapia ormonale) | 10000 UI | UD B |
| captopril (per l’aumento della pressione sanguigna) | compressa per somministrazione orale 12,5 mg | UD B |
| paracetamolo (antipiretico) | compressa per somministrazione orale 200 mg | UD B |
| etamsilato di sodio (per il sanguinamento) |
per la somministrazione orale per somministrazione endovenosa 2 ml |
UD B |
| colecalciferolo (per l'ipocalcemia) | Compresse da 500 mg | UD B |
Utilizzo di trasfusioni di concentrati piastrinici:
Indicazioni:
· presenza di emorragie potenzialmente letali.
Le trasfusioni di concentrato piastrinico devono sempre essere complementari alla terapia specifica per la PTI (IVIG e/o glucocorticoidi) e non devono essere utilizzate come monoterapia. Se la gravità del sanguinamento nella PTI è tale da richiedere la trasfusione di concentrato piastrinico, si raccomandano trasfusioni frazionate - ogni 6-8 ore. Nei casi particolarmente gravi si ricorre a trasfusioni “iperfrazionate” con piccole dosi di concentrato piastrinico: 1-2 dosi (0,7-1,4x10 11) ogni due ore. L'etamsilato e i farmaci antifibrinolitici vengono utilizzati come terapia emostatica aggiuntiva.
ATTENZIONE! In caso di sanguinamento renale la somministrazione di inibitori della fibrinolisi è controindicata.
Intervento chirurgico:
Splenectomia(UD-B)
Indicazioni per l'intervento:
· decorso ricorrente e grave della malattia per più di 6 mesi;
· pazienti di età superiore a 6 anni dopo precedente vaccinazione con Haemophilus influenzae di tipo b + S.pneumoniae + N.Meningitidis.
Controindicazioni all'intervento:
· bambini sotto i 6 anni;
· ITP primario.
Altri trattamenti: NO.
Terapia emostatica ausiliaria:
· sodio etamsilato 12,5% alla dose di 10-15 mg/kg;
· acido para-aminobenzoico - acido tranexamico: oltre i 12 anni alla dose di 20-25 mg/kg.
Indicazioni per la consultazione con specialisti:
· consultazione con uno specialista in malattie infettive - se si sospetta un processo infettivo;
· consultazione con un endocrinologo - se durante il trattamento si sviluppano disturbi endocrini;
· consultazione con un ostetrico-ginecologo - durante la gravidanza, metrorragia, menorragia, quando si prescrivono contraccettivi orali combinati;
· consultazione con altri specialisti ristretti - secondo le indicazioni.
Indicazioni per il trasferimento in terapia intensiva:
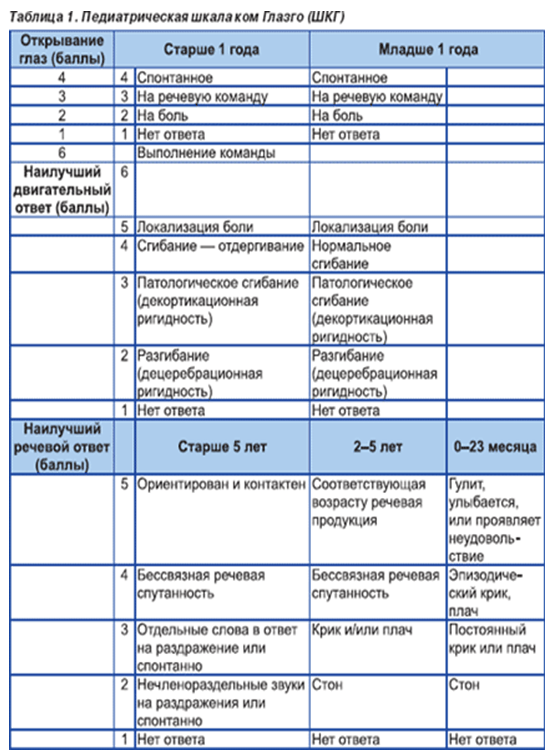
· assenza/compromissione della coscienza (scala di Glasgow); domanda n. 1
insufficienza cardiovascolare acuta (frequenza cardiaca inferiore a 60 o superiore a 200 al minuto);
· insufficienza respiratoria acuta (difficoltà respiratoria 2 - 3 gradi, frequenza respiratoria superiore a 50, diminuzione della saturazione inferiore all'88%, necessità di ventilazione meccanica);
Disturbi circolatori acuti (condizioni di shock);
· Pressione arteriosa sistolica, inferiore a 60/superiore a 180 (che richiede la somministrazione costante di farmaci vasoattivi);
· disturbi metabolici critici (elettroliti, acqua, proteine, equilibrio acido-base, chetoacidosi);
· osservazione intensiva e farmacoterapia intensiva, che richiedono un monitoraggio costante delle funzioni vitali;
· violazione dei sistemi di coagulazione del sangue e anticoagulanti.
Indicatori di efficacia del trattamento:
· dopo 4 settimane dall'inizio del trattamento, aumento delle piastrine superiore a 100x10 9 / l (75% dei pazienti con ITP).
· dopo la rimozione della milza - aumento del livello delle piastrine nel sangue periferico.
Ulteriore gestione
Ricerca di laboratorio:
· Una volta al mese nel primo anno di osservazione viene effettuato un emocromo con determinazione della conta piastrinica e conteggio manuale della formula leucocitaria (obbligatorio). Inoltre, a seconda delle condizioni cliniche e della stabilità del quadro ematologico;
· se indicato, si effettua l'analisi biochimica dinamica del sangue;
· test sierologici dei marcatori dell'HIV, dell'epatite B e C, effettuati 3 mesi dopo la dimissione dall'ospedale e 3 mesi dopo ogni trasfusione di emoderivati.
Condizioni per il trasferimento del paziente al luogo di residenza:
· il pediatra (ematologo pediatrico) del luogo di residenza si ispira alle raccomandazioni fornite dagli specialisti ospedalieri;
· la frequenza dell'esame di un paziente con ITP è una volta ogni 2-4 settimane nei primi 3 mesi di trattamento, poi a seconda delle condizioni cliniche e della dinamica ematologica, ma almeno una volta ogni 2 mesi.
Studi strumentali effettuato quando clinicamente indicato.
Ricovero ospedaliero
Indicazioni per il ricovero programmato:
Indicazioni per il ricovero d'urgenza:
Diminuzione del livello piastrinico nell'emocromo<50х10 9 /л.
· presenza di sindrome emorragica (sanguinamento dalle mucose del rinofaringe, del cavo orale, sanguinamento gastrointestinale, sanguinamento uterino).
Informazione
Fonti e letteratura
- Verbali delle riunioni della Commissione mista sulla qualità dei servizi medici del Ministero della Salute della Repubblica del Kazakistan, 2016
- 1) Ematologia pediatrica, 2015. A cura di A.G. Rumyantsev, A.A. Maschan, E.V. Zhukovskaya. Mosca. Gruppo editoriale "GEOTAR-Media" 2015 C – 656, C-251, tabella 6. 2) Linee guida pratiche basate sull'evidenza per la trombocitopenia immune dell'American Society of Hematology 2011 Cindy Neunert, Wendy Lim, Mark Crowther, Alan Cohen, Lawrence Solberg, Jr e Mark A. Crowther2011; 16:4198-4204 3) Standardizzazione dell'ITP, settembre 2006 IMBACH. 4) Fornire cure di emergenza, 2005. Algoritmo delle azioni in situazioni di emergenza: secondo le linee guida IMCI - OMS per la gestione delle malattie più comuni negli ospedali di livello primario, adattate alle condizioni della Repubblica del Kazakistan (OMS 2012). 5) ES. Il manuale "Trombocitopenia immune" 2011. 6) Tarantino & Buchanan, Hematol Oncol Clin North Am, 2004, 18:1301-1314. 7) Linee guida per la nutrizione parenterale amministrativa Canada 2010. 8) SIGN 104. Profilassi antibiotica in chirurgia.2014.
Informazione
Abbreviazioni utilizzate nel protocollo
| AG | ipertensione arteriosa; |
| INFERNO | pressione arteriosa; |
| ALLaT | alanina aminotransferasi |
| Al | aspartato aminotransferasi |
| IV | per via endovenosa |
| io sono | per via intramuscolare |
| VVID | terapia immunoglobulinica per via endovenosa ad alte dosi |
| HIV | virus dell'AIDS; |
| GGTP | gammaglutamil transpeptidasi; |
| IMCI | gestione integrata delle malattie infantili |
| ventilazione meccanica | ventilazione artificiale |
| E COSÌ VIA | trombocitopenia immunitaria |
| ELISA | test immunoassorbente collegato; |
| IFT | immunofenotipizzazione; |
| CT | TAC; |
| KSH | stato acido-base |
| LDH | lattato deidrogenasi; |
| Strutture sanitarie | istituzione medica |
| MDS | sindrome mielodisplasica; |
| ME | unità internazionali |
| MMF | micofenolato mofetina |
| risonanza magnetica | Risonanza magnetica |
| UAC | analisi del sangue generale |
| OAM | analisi generale delle urine; |
|
antiriciclaggio PNG |
leucemia mieloblastica acuta; emoglobinuria parossistica notturna; |
| ONMK | accidente cerebrovascolare acuto |
| PCR | reazione a catena della polimerasi; |
| VES | - velocità di sedimentazione eritrocitaria; |
| HSCT | trapianto di cellule staminali emopoietiche |
| USDG | Ecografia Doppler |
| FGDS | fibro-gastro-duadenoscopia |
| colpire | trombocitopenia immunitaria cronica |
| CMV | citomegalovirus |
| BH | frequenza respiratoria; |
| Frequenza cardiaca | frequenza cardiaca; |
| ECG | elettrocardiografia; |
| EcoCG | ecocardiografia; |
| Ig | immunoglobulina |
Elenco degli sviluppatori di protocolli con informazioni sulla qualifica:
1) Omarova Gulnara Erbosynovna - ematologa/oncologa pediatrica, filiale della Fondazione aziendale "UMC", "Centro scientifico nazionale per la maternità e l'infanzia", Astana.
2) Tastanbekova Venera Bulatovna - ematologa/oncologa pediatrica, filiale della Fondazione aziendale “UMC”, “Centro scientifico nazionale per la maternità e l'infanzia”, Astana.
3) Umirbekova Balzhan Bolatovna - ematologo/oncologo pediatrico, filiale della Fondazione aziendale "UMC", "Centro scientifico nazionale per la maternità e l'infanzia", Astana.
4) Omarova Kulyan Omarovna - Dottore in Scienze Mediche, Professore, Centro Nazionale di Pediatria e Chirurgia Infantile, Almaty.
5) Manzhuova Lyazzat Nurpapaevna - Candidato di scienze mediche, capo del dipartimento di oncologia n. 1, Centro nazionale di pediatria e chirurgia infantile, Almaty.
6) Mira Maratovna Kalieva - Candidata di scienze mediche, professore associato del Dipartimento di farmacologia clinica e farmacoterapia di KazNMU dal nome. S. Asfendiyarova.
Indicazione di assenza di conflitti: NO.
Elenco dei revisori: Kemaykin Vadim Matveevich - ematologo della più alta categoria di qualifica, candidato in scienze mediche, capo ematologo freelance, oncoematologo del Ministero della sanità e dello sviluppo sociale della Repubblica del Kazakistan.
Allegato 1
Files allegati
Attenzione!
- Automedicando, puoi causare danni irreparabili alla tua salute.
- Le informazioni pubblicate sul sito web MedElement non possono e non devono sostituire una consultazione faccia a faccia con un medico. Assicurati di contattare una struttura medica se hai malattie o sintomi che ti preoccupano.
- La scelta dei farmaci e il loro dosaggio devono essere discussi con uno specialista. Solo un medico può prescrivere il medicinale giusto e il suo dosaggio, tenendo conto della malattia e delle condizioni del corpo del paziente.
- Il sito web MedElement è esclusivamente una risorsa informativa e di riferimento. Le informazioni pubblicate su questo sito non devono essere utilizzate per modificare senza autorizzazione le prescrizioni del medico.
- Gli editori di MedElement non sono responsabili per eventuali lesioni personali o danni alla proprietà derivanti dall'uso di questo sito.
Trombocitopenia- le cause più comuni di aumento del sanguinamento nei bambini, mentre la componente piastrinica dell'emostasi viene interrotta
Con il termine “porpora” (lat. porpora - preziosa pittura antica di colore viola scuro) si intendono piccole emorragie nello spessore della pelle o sulle mucose. Le piccole emorragie punteggiate sono chiamate petecchie, le emorragie più grandi sono chiamate ecchimosi (emorragie puntiformi).
Si distinguono le seguenti forme di trombocitopenia:
viola:
1. Primaria - malattia di Werlhoff idiopatica, forme congenite di trombocitopatie (oltre 60 sindromi e malattie).
2. Le forme secondarie (acquisite) si verificano in molte malattie: sindrome DIC, leucemia, lupus eritematoso sistemico, ecc.
L'incidenza della porpora trombocitopenica all'anno è di 4,5-7,5 su 100.000, di cui il 47% si verifica in porpora trombocitopenica idiopatica primaria.
Per tutti i gruppi di malattie, i segni caratteristici sono la trombocitopenia e la sindrome emorragica.
PURPURA TROMBOCITOPENICA IDIOPATICA (ITP)
Porpora trombocitopenica idiopatica (malattia di Werlhof) - una malattia caratterizzata da una tendenza al sanguinamento ed espressa nell'inferiorità quantitativa e qualitativa della componente piastrinica dell'emostasi. In termini di frequenza delle malattie, la porpora trombocitopenica è al primo posto tra la diatesi emorragica.
La malattia fu descritta da Werlhof nel 1735 con il nome di “malattia emorragica maculata”.
L'incidenza della porpora trombocitopenica è di 1 su 10.000 bambini. In età prescolare La malattia di Werlhof viene registrato nei ragazzi e nelle ragazze con la stessa frequenza e dopo 10 anni nelle ragazze - 3 volte più spesso.
La porpora trombocitopenica è causata da trombocitopatia ereditaria e alterazioni immunopatologiche.
I principali fattori di rischio per lo sviluppo della porpora trombocitopenica:
Predisposizione ereditaria (ereditarietà autosomica dominante sotto forma di carenza piastrinica qualitativa);
Lesioni mentali e fisiche;
Reazioni immunopatologiche a vaccini, sieri, antibiotici, salicilati, sulfamidici, ecc.;
Infezioni virali e batteriche;
Iperinsolazione o ipotermia;
Fattori ambientali avversi. Il meccanismo di sviluppo della porpora trombocitopenica.
Il meccanismo di sviluppo della porpora trombocitopenica non è ancora del tutto chiaro.
Grande importanza è attribuita al processo immunopatologico, che è evidenziato dallo sviluppo della porpora 2-3 settimane dopo una malattia virale-batterica e dal rilevamento di anticorpi antipiastrinici nel sangue di pazienti che hanno sofferto di porpora trombocitopenica. Inoltre, viene migliorata la funzione di formazione delle piastrine del midollo osseo, il che porta ad un aumento del numero di cellule giovani immature (megatrombociti) nel sangue. La loro durata di vita si riduce a diverse ore, invece dei normali 7-10 giorni.
Nella malattia di Werlhof, il sanguinamento è causato non solo dalla trombocitopsnia, ma anche da una violazione secondaria della resistenza della parete vascolare (a causa della perdita della funzione angiotrofica delle piastrine) e da una diminuzione della contrattilità vascolare (a causa di una diminuzione del livello di grigiastro nel sangue - un vasocostrittore contenuto nelle piastrine). Il sanguinamento è mantenuto dal fatto che non si forma un coagulo completo a causa di una violazione della sua retrazione.
La malattia inizia in modo acuto, 2-3 settimane dopo la malattia virale-batterica.
Le principali manifestazioni cliniche della porpora trombocitopenica.
I sintomi dell'intossicazione sono moderati, la temperatura è bassa, la debolezza e il malessere sono preoccupanti;
Più spesso di notte, a causa di lievi ferite o compaiono spontaneamente sanguinamento nella pelle, Per emorragie cutanee (porpora) L'eruzione cutanea è caratterizzata da polimorfismo (da grandi emorragie - ecchimosi a piccoli punti - pe-tech), assenza di dolore e localizzazione asimmetrica dell'eruzione cutanea, spesso sulle estremità e sulla superficie anteriore del corpo, inadeguatezza delle lesioni e gravità delle emorragie (un piccolo colpo o anche un tocco provoca estese ecchimosi sottocutanee), eruzione policroma - il colore delle emorragie cambia gradualmente dal cremisi al blu-verde e al giallo, attraversando tutte le fasi della fioritura del livido (a causa della conversione dell'emoglobina in bilirubina); con emorragie multiple la pelle diventa simile alla pelle di un leopardo, completamente scolorito sch l'assorbimento delle ecchimosi avviene dopo 3 settimane;
emorragie nelle mucose, in questo caso possono essere colpite la sclera (“lacrime di sangue”) e la retina degli occhi, le tonsille, il palato molle e duro, la parete posteriore della faringe e il timpano dell'orecchio medio;
altri tipi di sanguinamento più comuni: nasale, gastrointestinale, dalle gengive o dall'alveolo di un dente estratto, nelle ragazze adolescenti è possibile un grave sanguinamento uterino durante le mestruazioni (80%), che dura fino a 2-4 settimane ed è difficile da fermare;
violazione della resistenza della parete vascolare(rilevato da un laccio emostatico e da un sintomo di pizzicamento, un test del polsino o della coppa);
cambiamento nell'emogramma: una diminuzione del numero di piastrine nel sangue - fino alla loro completa scomparsa, una diminuzione della retrazione del coagulo sanguigno (meno del 60%), un aumento del tempo di sanguinamento secondo il metodo Duque a 20 minuti o più (normalmente - non più meno di 4 minuti).
Va ricordato che l'emorragia nella sclera degli occhi può essere il primo segno emorragie nel cervello.
I principali segni clinici di emorragia cerebrale:
Forte mal di testa, vertigini;
Convulsioni, vomito;
Sintomi neurologici patologici: angolo cadente della bocca, asimmetria della fessura palpebrale, disturbi del linguaggio, movimenti attivi, ecc. Metodi diagnostici di laboratorio:
1. Esame del sangue clinico.
2. Coagulogramma.
3. Puntura del midollo osseo.
4. Analisi generale delle urine.
5. Esame del sangue immunologico e biochimico.
6. Reazione di Coombs, test di emoagulazione di aggregazione. Principi di base del trattamento.
1. Ricovero obbligatorio.
2. Rigoroso riposo a letto fino alla stabilizzazione dell'emogramma (ripristino del livello fisiologico minimo delle piastrine).
3. Dieta ipoallergenica.
4. Agenti emostatici generali: somministrazione endovenosa di acido epsilon-aminocaproico.
5. Agenti emostatici locali: spugna emostatica o di gelatina, film di fibrina, tamponi con acido epsilon-aminocaproico o perossido di idrogeno, trombina.
6. Agenti che migliorano l'aggregazione piastrinica: dicinone, pantotenato di calcio, adroxon, etamsylate, ecc.
7. Corsi di farmaci corticosteroidi: prednisolone (per sanguinamento pesante, prolungato ripetuto).
8. Immunoglobuline (flebo endovenoso per 4 giorni).
9. Fitoterapia: achillea, ortica, fragole, rosa canina, peperoncino, arachidi, ecc.
10. Terapia vitaminica: preparati di vitamine C, P, gruppo B.
11. In caso di grave sanguinamento massiccio che minaccia la vita, la splenectomia viene eseguita sullo sfondo della terapia con corticosteroidi. L'operazione porta al recupero pratico.
12. Gli immunosoppressori vengono prescritti se non si riscontra alcun effetto da altri tipi di terapia. Previsione.
Generalmente favorevole alla vita. In rari casi, un sanguinamento grave può provocare la morte. Prevenzione.Prevenzione primaria:
1. Consulenza genetica per l'identificazione della predisposizione ereditaria.
2. Proteggi il bambino dalle infezioni virali e batteriche.
3. Prestare attenzione durante la somministrazione di vaccini, sieri, antibiotici, salicilati, sulfamidici, ecc.
4. Proteggere da lesioni mentali e fisiche.
5. Evitare l'iperinsolazione e l'ipotermia.
6. Effettuare tempestivamente la sanificazione dei focolai cronici di infezione.
Prevenzione secondaria(finalizzato a prevenire la recidiva della malattia):
1. Prevenzione dei vaccini - secondo un calendario individuale.
2. Per prevenire il sanguinamento uterino, si consiglia di somministrare acido aminocaproico alle ragazze adolescenti dal 1° al 5° giorno delle mestruazioni.
3. I bambini guariti dalla malattia sono esentati dall'educazione fisica nel gruppo principale e dalla partecipazione alle competizioni per 5 anni.
4. Si raccomanda ai convalescenti di porpora trombocitopenica di effettuare esami del sangue di controllo con conteggio del numero delle piastrine e determinazione del tempo di sanguinamento secondo Duque: nei primi 3 mesi dopo la malattia - 2 volte al mese, poi - 1 volta al mese per un anno, successivamente - 2 volte l'anno .
5. I bambini che hanno sofferto di porpora trombocitopenica sono soggetti a osservazione clinica da parte di un pediatra ed ematologo per 5 anni.
EMOFILIA
Emofiliaè una malattia correlata a coagulopatie ereditarie, a trasmissione recessiva,
Tipo legato all'X, caratterizzato da sanguinamento difficile da arrestare causato da una deficienza nell'attività dei fattori plasmatici VIII, IX o XI del sistema di coagulazione del sangue.
Il termine “emofilia” fu proposto da Schonlein nel 1820. Egli identificò l'emofilia come una malattia indipendente e ne descrisse i segni clinici caratteristici.
La frequenza dell'emofilia A, secondo l'OMS, è di 0,5 - 1 caso ogni 10.000 neonati, l'emofilia B è 5 volte meno comune. Ancora più rara è l'emofilia C, che rappresenta l'1-2% dei casi di tutte le coagulopatie ereditarie. ,
Esistono 3 tipi di emofilia (a seconda della carenza di fattori della coagulazione del sangue):
1. Emofilia A caratterizzato da deficit del fattore VIII - globulina antiemofila.
2. Emofilia B accompagnato da un disturbo della coagulazione del sangue dovuto alla mancanza del fattore IX, un componente della tromboplastina plasmatica.
3. Emofilia C - la forma più rara associata a deficit del fattore XI, precursore della tromboplastina plasmatica.
Il meccanismo di sviluppo dell'emofilia.
Per l'emofilia A e B L'ereditarietà della malattia avviene secondo un carattere recessivo legato al sesso. Ad esserne colpiti sono soprattutto gli uomini. La malattia si trasmette da nonno a nipote attraverso la figlia, conduttrice. Il cromosoma patologico è ereditato da un padre emofiliaco e dalle figlie. Allo stesso tempo, le figlie non soffrono di emofilia, poiché il cromosoma X alterato (del padre) è compensato da un cromosoma X completo (della madre).
Emofilia C ha una trasmissione di tipo autosomico recessivo, cioè sono colpite persone di entrambi i sessi e la tendenza al sanguinamento è trasmessa sia dalle donne che dagli uomini.
Una manifestazione caratteristica dell'emofilia è la sindrome emorragica.
Principali manifestazioni cliniche dell'emofilia.sanguinamento massiccio e tendenza a diffondersi con formazione di estesi ematomi(sottocutaneo e intermuscolare profondo e doloroso), il cui riassorbimento avviene lentamente, poiché il sangue versato rimane liquido per lungo tempo;
dolore intenso nell'area di ematomi estesi dovuti alla compressione dei nervi periferici e dei grandi vasi, a seguito dei quali possono svilupparsi paralisi o cancrena;
sanguinamento delle articolazioni (emartro), Di norma, sono colpite le articolazioni grandi (anca, ginocchio, gomito, caviglia), aumentano di volume, sono molto dolorose, la pelle sopra di esse è calda, emorragie ripetute portano alla loro rigidità (anchilosi) e deformazioni (si formano coaguli fibrinosi sulla capsula articolare e sulla cartilagine, che successivamente si trasformano in tessuto connettivo);
sanguinamento prolungato dalle mucose del naso, delle gengive, della bocca, meno spesso dal tratto gastrointestinale, dai reni, dall'alveolo dei denti durante l'estrazione o dopo la tonsillectomia, manipolazioni invasive, soprattutto dopo iniezioni intramuscolari;
caratterizzato da sanguinamento tardivo(di solito si verificano qualche tempo dopo l'infortunio);
cambiamento nell'emogramma: prolungamento del tempo di coagulazione del sangue capillare e venoso, rallentamento del tempo di ricalcificazione, ridotta formazione di tromboplastina, ridotto consumo di protrombina, ridotta quantità di uno dei fattori antiemofilici (VIII, IX, XI).
Caratteristiche del decorso dell'emofilia nei neonati:
Dopo la nascita si riscontrano cefaloematomi e/o segni di emorragia intracranica;
Poche ore dopo la nascita si rilevano emorragie spontanee nella pelle e nel tessuto sottocutaneo;
Sanguinamento prolungato dal cordone ombelicale legato.
Nel primo anno di vita Durante la dentizione può verificarsi sanguinamento.
Ma la malattia appare più spesso dopo un anno, quando il bambino inizia a camminare e aumenta il rischio di lesioni.
L'emofilia C è caratterizzata da un decorso più lieve.
Principi di base del trattamento.
1. Rigoroso riposo a letto durante l'esacerbazione della malattia fino alla stabilizzazione della condizione.
2. Terapia sostitutiva: per l'emofilia A è indicata la somministrazione di globulina antiemofila concentrata (crioprecipitato) alla dose di 15-50 UI/kg (la sua emivita è di 4-8 ore), pertanto sono necessarie infusioni ripetute in caso di ematomi massivi o emorragie; per l'emofilia B e C si utilizza plasma nativo concentrato di 150-300 unità (i fattori IX e XI sono abbastanza stabili); Per il trattamento dei pazienti affetti da emofilia B, il complesso farmaco PPSB è più efficace.
3. Per l'emostasi locale: film di fibrina, spugna emostatica, trombina, latte umano fresco ricco di tromboplastina.
4. Per emartro - immobilizzazione dell'arto in posizione fisiologica (per 2-3 giorni), raffreddore locale.
5. In caso di emorragia massiva dell'articolazione: puntura immediata dell'articolazione con aspirazione del sangue e iniezione di una sospensione di idrocortisone nella cavità articolare.
6. In futuro sono indicati un leggero massaggio dei muscoli dell'arto interessato, un uso attento di esercizi terapeutici e procedure fisioterapeutiche.
7. Se si sviluppa anchilosi, è necessario un trattamento chirurgico.
8. Corsi di preparazioni corticosteroidi: prednisolone (per sanguinamento pesante, prolungato ripetuto).
9. Con lo sviluppo dell'anemia postemorragica: ferroterapia.
Prevenzione.
1. Consulenza medica e genetica tempestiva al paziente e ai familiari.
2. Osservare le precauzioni durante gli interventi invasivi: vengono eseguite solo infusioni endovenose, sono vietate eventuali iniezioni intramuscolari e sottocutanee.
3. Effettuare una terapia sostitutiva prima di qualsiasi intervento chirurgico, compresi quelli dentali.
4. Per il minimo sanguinamento: somministrazione di farmaci antiemofilici concentrati. 5. Condurre la profilassi vaccinale secondo un calendario individuale e solo in centri antiemofilici specializzati.
| 6. |
Esentare i bambini in età scolare dall'educazione fisica (per evitare lesioni fisiche). Esami del sangue di controllo obbligatori dopo gli infortuni.
8. Non è consentito assumere acido acetilsalicilico nel trattamento di malattie concomitanti.
9. Monitoraggio costante da parte di un ematologo in un centro antiemofilico.
Previsione.
La malattia è incurabile.
La prognosi dipende dalla gravità della malattia, dalla tempestività e dall'adeguatezza della terapia etiotropica.
Le emorragie nel cervello e nelle meningi possono portare alla morte o a gravi danni organici al sistema nervoso centrale. Processo infermieristico nelle sindromi emorragiche.
Informare i genitori e il paziente sulle possibili cause della malattia, sulle sue manifestazioni, sulle misure preventive, sui principi di base del trattamento e sulle possibili complicanze.
Individuare tempestivamente i problemi reali e potenziali ed i bisogni vitali del paziente e dei suoi familiari.
Possibili problemi del paziente:
Cattiva salute, aumento della fatica;
Dolore nell'area delle emorragie, delle articolazioni, dell'addome;
Attività fisica e motoria compromessa;
Cambiamento nell'aspetto;
L’incapacità del bambino di affrontare autonomamente le difficoltà derivanti dalla malattia;
Paura del ricovero in ospedale, della manipolazione;
Separazione a lungo termine dai propri cari e dai coetanei, grave reazione al ricovero in ospedale;
Diminuzione dell'attività cognitiva, squilibrio psico-emotivo;
Disadattamento sociale;
Alto rischio di disabilità;
Difficoltà nella scelta di una professione;
Alto rischio di complicanze. Possibili problemi per i genitori:
Mancanza di conoscenza della malattia e della cura;
Cambiare lo stereotipo della vita alla nascita di un bambino affetto da emofilia;
Scarsa comprensione dei bisogni del bambino;
Mancanza di fiducia nel trattamento fornito;
Costante stress psico-emotivo e paura per l'esito della malattia;
Valutazione inadeguata delle condizioni del bambino;
Iperprotezione, assecondando i capricci del bambino;
Senso di colpa davanti al bambino, impotenza, depressione;
Cambiamenti nei rapporti familiari, ecc. Intervento infermieristico.
Convincere i genitori e il bambino, se l'età e le condizioni lo consentono, della necessità di ricovero in un reparto specializzato nel periodo acuto della malattia per ricevere cure mediche qualificate.
Fornire assistenza nell'organizzazione del ricovero.
Fornire al paziente il riposo a letto (durante un periodo acuto o durante un'esacerbazione della malattia) e la pace psico-emotiva, proteggendolo da preoccupazioni e manipolazioni inutili. È necessario espandere gradualmente il regime motorio, 2 settimane dopo la fine delle eruzioni cutanee emorragiche. Criteri per l'espansione della modalità: miglioramento del benessere e delle condizioni generali, normalizzazione dei parametri di laboratorio, assenza di complicanze. Quando si passa al regime generale, è necessario condurre un test ortostatico (il bambino cammina per 1-2 ore, se non compaiono nuove eruzioni cutanee il giorno successivo, il regime può essere ampliato).
Monitorare le funzioni vitali: temperatura, polso, pressione sanguigna, condizioni della pelle e delle mucose, natura delle funzioni fisiologiche, ecc. Utilizzare un catetere succlavio per somministrare farmaci antiemofilici concentrati e, soprattutto, quando si eseguono altri interventi invasivi. Evitare iniezioni intramuscolari e sottocutanee!
Interagire in squadra con gli specialisti, coinvolgere i genitori e il bambino nel processo di cura (se l'età e le condizioni lo consentono). Insegnare ai genitori a fornire assistenza tempestiva di emergenza in caso di complicazioni: in caso di dolore, valutare la sindrome del dolore su una scala a 10 punti per un efficace sollievo dal dolore; durante lo sviluppo del bambino emartrosi fornirgli un rigoroso riposo a letto e immobilizzare l'arto in una posizione fisiologica (per 2-3 giorni), applicare il freddo localmente; in caso di epistassi: calmare il bambino, farlo sedere, inclinare la testa in avanti, applicare freddo sul ponte del naso e sulla nuca, premere le ali del naso sul setto nasale, eseguire il tamponamento nasale anteriore con turunde imbevute di una soluzione al 3% di perossido di idrogeno o agenti emostatici. Consigliare ai genitori di avere nel frigorifero una scorta di agenti emostatici locali: pellicola di fibrina, spugna emostatica, trombina, ecc.
Fornire al bambino un'alimentazione adeguata. Eliminare gli allergeni obbligati dalla dieta: cacao, cioccolato, agrumi, uova, frutta e verdura di colore rosso-arancio. Limite nella dieta: sale, estratti. Si consiglia di introdurre inoltre nella dieta: succhi diluiti, bevande alla frutta, composte di frutta secca, acidophilus, ricotta, bio-yogurt, bio-kefir, arachidi, spinaci, aneto, ortica. Nel caso della forma renale di vasculite emorragica, nei primi 5-7 giorni di malattia viene prescritta la dieta n. 7 (tavola priva di sale con limitazione delle proteine animali, escludendo dalla dieta carne e pesce), nel caso della sindrome addominale - dieta n. 1.
Fornire consigli ai genitori sulla fitoterapia e sulla selezione delle erbe (preparati emostatici), insegnare loro la tecnologia di preparazione. La collezione comprende: erba di San Giovanni, ortica, achillea, fragole, pepe d'acqua, seta di mais, rosa canina, borsa del pastore, aronia
sorbo, foglia di ribes nero, labbro leporino inebriante. La proporzione delle erbe nella collezione è 1:1. Metodo di preparazione: preparare 1 cucchiaio della raccolta con un bicchiere di acqua bollente, lasciare agire per 10-15 minuti, assumere 1/2-1/3 bicchiere 2-3 volte al giorno 20 minuti prima dei pasti, per 2 mesi con una pausa di un mese.
Organizzare l'attività cognitiva del bambino: tenerlo impegnato nella lettura dei suoi libri preferiti, concedergli momenti di svago interessanti (calma, giochi divertenti, disegno, ascolto della radio, visione di programmi televisivi per bambini, ecc.).
Fin dalla tenera età, è necessario introdurre elementi di cautela quando si svolgono giochi all'aperto, ma non si dovrebbe mostrare maggiore preoccupazione per non provocare un senso di inferiorità nel bambino. Fornire supporto psicologico al paziente e ai suoi parenti: convincere i genitori della necessità di fiducia reciproca tra loro e il bambino, instillare costantemente in lui che è sano da un punto di vista mentale e fisico, presentarlo a coetanei che hanno una malattia simile , instillare in lui fiducia nelle sue capacità.
Elaborare insieme ad uno psicologo un programma di psicocorrezione per un corretto orientamento sociale e professionale. A casa è necessario creare le condizioni per lo sviluppo delle inclinazioni al lavoro mentale (in caso di emofilia, orientare costantemente il bambino verso il lavoro a casa, che elimini il più possibile traumi e attività fisica). Consigliare ai genitori di creare rapporti paritari in famiglia, trattare tutti i bambini allo stesso modo, evitare l'iperprotezione di un bambino malato, correggere il suo comportamento in modo tempestivo, discutere i suoi sentimenti con lui più spesso, evitando frasi che evocano simpatia.
Convincere i genitori della necessità del monitoraggio dinamico del bambino da parte di medici - ematologo, pediatra e altri specialisti secondo le indicazioni al fine di monitorare la condizione e i parametri ematologici e adattare la terapia
PROCESSO INFERMIERISTICO NELLA LEUCEMIA ACUTA
Leucemia acutaè una malattia maligna del sistema ematopoietico e linfoide con localizzazione primaria del processo patologico nel midollo osseo e successiva metastasi ad altri organi.
Il termine “leucemia” fu proposto per la prima volta nel 1921 da Ellerman. Gli scienziati domestici hanno dato un grande contributo allo studio della leucemia: I. A. Kassirsky, A. I. Vorobyov, Yu. I. Lorie, A. F. Tur, N. S. Kislyak, L. A. Makhonova, I. V. Koshel .
L'incidenza della leucemia infantile è di 2-5 su 100.000 bambini. Il picco di incidenza della malattia si verifica tra i 2 e i 5 anni di età. I ragazzi soffrono di leucemia 1,5 volte più spesso delle ragazze. Nei bambini, la leucemia acuta viene diagnosticata più spesso di qualsiasi altro tumore.
È consuetudine distinguere le seguenti forme di leucemia acuta nell'infanzia:
1. Leucemia linfoide acuta (rappresenta il 75%), divisa in tre varianti morfologiche (cellule Li - L3).
2. La leucemia mieloblastica acuta (15-17%), è divisa in sei varianti morfologiche (cellule M, - Mb) - questa forma è più spesso osservata nell'adolescenza.
3. Leucemia acuta indifferenziata (5-9%), con tipi cellulari (Lo - Mo);
4. Leucemia mieloide cronica (1-3%).
Negli ultimi decenni si è registrato un aumento dell’incidenza della leucemia, tuttavia sono stati ottenuti alcuni successi nel trattamento della malattia e nel miglioramento della qualità della vita dei pazienti affetti da leucemia. Fattori di rischio per lo sviluppo della leucemia:
Predisposizione ereditaria (la conferma è la leucemia familiare e la leucemia nei gemelli identici);
Aberrazioni cromosomiche (l'anomalia cromosomica più comune è aneuploidia- cambiamento nel numero di cromosomi, quindi nella leucemia mieloide cronica una coppia di cromosomi è quasi 2 volte inferiore al normale; nella sindrome di Down il rischio di sviluppare leucemia acuta aumenta di 20-30 volte);
Caratteristiche costituzionali del corpo (iperplasia del timo, aumento dello sviluppo fisico, grave macrosomia facciale e altri stimmi di disembriogenesi);
L'impatto di fattori chimici esogeni sul corpo: benzene, steroidi (ormoni sessuali, acidi biliari, colesterolo), composti azotati, prodotti metabolici del triptofano, insetticidi;
Esposizione alle radiazioni ionizzanti (un aumento significativo del numero di malattie nelle aree colpite dalle radiazioni: Hiroshima, Nagasaki, Chernobyl, Chelyabinsk, ecc.).
Il meccanismo di sviluppo della leucemia.
Esiste una teoria virale sull'origine della leucemia: sotto l'influenza di agenti cancerogeni chimici, radiazioni e altri influssi ambientali avversi, si attivano virus oncogeni (permanentemente presenti nel corpo), che causano la proliferazione patologica di cellule indifferenziate, in combinazione con l'azione immunologica inerzia del corpo rispetto all'agente leucemico.
La teoria monoclonale dello sviluppo della leucemia è generalmente accettata. Secondo questa teoria, le cellule leucemiche sono un clone, la progenie di una cellula cellula mutata che ha cessato la sua differenziazione in uno dei primi livelli di maturazione. Le mutazioni cellulari avvengono quasi continuamente; in media, una cellula muta ogni ora. Nelle persone sane, il sistema immunitario si attiva, reagendo a queste cellule come se fossero estranee ed eliminandole. Nella leucemia acuta, le informazioni vengono interrotte
formazione e differenziazione cellulare e la loro liberazione dal controllo dei fattori regolatori. Un segno caratteristico della leucemia acuta è l'aumento del numero dei blasti nel midollo osseo. Cause di danni al midollo osseo soppressione della normale emopoiesi dovuto allo spostamento delle cellule normali. Le cellule tumorali si diffondono attraverso il sistema ematopoietico; sono in grado di oltrepassare i vasi sanguigni in tutti gli organi e tessuti dove si sviluppano infiltrati leucemici (metastasi). Molto spesso, l'ematopoiesi patologica si verifica dove esisteva nel periodo embrionale: nella milza, nei linfonodi e nel fegato.
Di conseguenza, lo sviluppo della leucemia è associato agli effetti combinati di fattori mutageni sfavorevoli e alla formazione di reazioni immunopatologiche nel corpo.
Le manifestazioni cliniche dipendono dal grado di inibizione della normale emopoiesi e dalla gravità dell'infiltrazione leucemica degli organi.
Ci sono quattro periodi della malattia:
1. Principiante.
2. Pieno sviluppo.
3. Remissioni.
4. Terminale.
Principali manifestazioni cliniche della leucemia acuta.
1. Nel periodo iniziale:
I sintomi di intossicazione sono espressi: aumento dell'affaticamento, diminuzione dell'appetito, disturbi del sonno, malessere, letargia, febbre leggera;
La pelle e le mucose diventano pallide, sulla pelle compaiono piccole emorragie.
2. Nel periodo di pieno sviluppo caratteristica sin
dromi:
sindrome osteoarticolare: il dolore compare gradualmente nelle ossa tubulari (femore e tibia), associato a infiltrazione leucemica del midollo osseo;
sindrome emorragica(uno dei segni più eclatanti della leucemia acuta), con emorragie notate
nella pelle, nelle mucose, nelle cavità articolari, nel cervello, può verificarsi sanguinamento dal naso, dalle gengive, dal tratto gastrointestinale, dai reni;
sindrome necrotica ulcerativa: danno alla pelle e alle mucose causato dall'infiltrazione leucemica dei tessuti e dei vasi sanguigni, dalla presenza di emorragie e successivamente dall'aggiunta di un'infezione virale-batterica;
linfoadenopatia sistemica(Linfonodi ingrossati): cervicali, sottomandibolari, ascellari, inguinali, che acquistano una densa consistenza elastica, sono indolori e non fusi con il tessuto circostante;
sindrome cardiovascolare: tachicardia, suoni cardiaci ovattati, soffio funzionale, i confini del cuore sono espansi;
sindrome epato-lienale;
sindrome ematologica: nel sangue periferico compaiono cellule immature (potente) cellule, anemia, trombocitopenia, variazione del numero dei leucociti da elevata leucocitosi a forte diminuzione, successivamente potrebbe esserci una mancanza di forme transitorie tra cellule giovani e mature (gape leucemico), c'è un aumento
nel midollo osseo punteggiato: vengono rilevate esplosioni
cellule, eritro e trombopoiesi sono inibite.
Successivamente, con il progredire della malattia, il processo leucemico colpisce organi non emopoietici:
tratto gastrointestinale(sindrome dispeptica e addominale);
sistema genito-urinario(reni ingrossati, ematuria, insufficienza renale, testicoli ingrossati nei ragazzi, ovaie nelle ragazze);
sistema nervoso centrale (sindrome neuroleucemia - mal di testa, nausea, vomito, disturbi della coscienza, convulsioni), possono svilupparsi neurite e paralisi.
Durante questo periodo viene spesso espresso Complesso di sintomi di Mikulicz: il viso diventa gonfio a causa di un aumento simmetrico delle ghiandole sottomandibolari, parotidi, periorbitali e salivari;
La dimensione del fegato e della milza aumenta.
Durante la leucemia ci sono diverse fasi:
Fase I- primo attacco della malattia (il periodo che va dall'esordio delle manifestazioni cliniche fino all'ottenimento dell'effetto della terapia).
Fase II - remissione della malattia.
Esistono remissioni complete e incomplete:
Con remissione clinica ed ematologica completa: non sono presenti sintomi clinici e il mielogramma mostra meno del 10% di blasti e meno del 20% di linfociti.
In caso di remissione clinica ed ematologica incompleta: le manifestazioni cliniche e i parametri dell'emogramma sono normalizzati, ma non vi è alcuna normalizzazione del mielogramma.
Fase III- recidiva della malattia dovuta al ritorno del processo leucemico. Più spesso, una recidiva della malattia inizia con la comparsa di focolai extramidollari di infiltrazione leucemica nei testicoli, nel sistema nervoso, nei polmoni, sullo sfondo della normale emopoiesi. Sintomi meno gravi durante il periodo di recidiva della leucemia acuta sono associati a un trattamento complesso continuo che inibisce lo sviluppo del processo leucemico.
Informazioni correlate.