Eziologia della distrofia granulare. Distrofia renale. Impostazione del tema di destinazione
Alterazione(dal latino alteratio – cambiamento) o danno sono cambiamenti strutturali nelle cellule e nei tessuti del corpo che si verificano sotto l'influenza di fattori esogeni ed endogeni. Allo stesso tempo, il metabolismo, la funzione e l’attività vitale dei tessuti e degli organi vengono interrotti. Le cause dell'alterazione possono essere disturbi circolatori, agenti fisici e sostanze chimiche, agenti infettivi, reazioni immunopatologiche, fattori genetici e uno squilibrio di sostanze necessarie alla cellula (solitamente dovuto a disturbi nutrizionali). L'entità del danno alle cellule e ai tessuti dipende dal tipo e dalla durata d'azione del fattore patogeno, dalle caratteristiche morfo-funzionali del macroorganismo. L'alterazione può verificarsi a livello ultrastrutturale, cellulare, tissutale e di organo.
Nella patogenesi del danno cellulare e tissutale, i fattori ipossici associati ad un insufficiente apporto di ossigeno ai tessuti (ad esempio, quando il flusso sanguigno arterioso è ostruito) svolgono un ruolo importante. Per l'ipossia
La fosforelazione ossidativa si arresta e la sintesi di ATP diminuisce, la glicolisi viene attivata. Il gonfiore acuto (edema) della cellula si verifica a causa di una violazione della pressione osmotica e dell'accumulo di metaboliti (fosfati, lattato, ecc.) Nel citoplasma, le cisterne del reticolo endoplasmatico si gonfiano, i mitocondri si espandono, le membrane plasmatiche sono danneggiate, compreso il lisosoma membrane, che porta al rilascio dei loro enzimi e alla rottura dei componenti cellulari.
Il danno cellulare può essere causato da radicali liberi dell’ossigeno, fattori chimici e radiazioni ionizzanti; si sviluppa durante l’invecchiamento cellulare. In questo caso si verifica la perossidazione dei lipidi di membrana, che può portare alla distruzione della membrana plasmatica e degli organelli. Un ruolo significativo è svolto anche dalla trasformazione ossidativa delle proteine, che aumenta la distruzione degli enzimi chiave attraverso proteasi neutre e il danno al DNA cellulare da parte dei radicali liberi.
I danni possono essere causati da sostanze chimiche che possono agire direttamente sulle molecole cellulari e sugli organelli (ad esempio, composti idrosolubili del cloruro di mercurio).
L'alterazione è rappresentata da due processi patologici: distrofia e necrosi, che possono essere fasi successive.
Distrofia(dal greco dis – prefisso che denota disordine e trofeo – nutrizione) è un processo patologico complesso, che si basa su disordini metabolici che portano a cambiamenti strutturali. In termini morfologici, durante la degenerazione di cellule e tessuti, compaiono sostanze normalmente assenti o contenute in piccole quantità, oppure scompaiono da cellule e tessuti sostanze ad esse inerenti.
La distrofia si basa su una violazione di un complesso di meccanismi che assicurano il metabolismo e la conservazione delle strutture cellulari e tissutali, come la funzione regolatrice del sistema nervoso ed endocrino, i disturbi dell'afflusso sanguigno e della circolazione linfatica, nonché i disturbi dell'autoregolazione cellulare , che porta all'interruzione dei processi enzimatici nella cellula.
I disturbi congeniti dei sistemi enzimatici (enzimepatie) sono la causa di un ampio gruppo di malattie ereditarie - thesaurismi (malattie da accumulo), in cui, a causa dell'assenza di qualsiasi enzima o della sua struttura errata, qualsiasi parte del metabolismo viene bloccata e nelle cellule compaiono sostanze anormali.
Si distinguono i seguenti meccanismi morfogenetici che portano allo sviluppo della distrofia:: infiltrazione, decomposizione (fanerosi), sintesi perversa, trasformazione.
Infiltrazione – si tratta dell'eccessiva penetrazione di prodotti metabolici dal sangue e dalla linfa nelle cellule o nella sostanza intercellulare con il loro successivo accumulo. Un tipico esempio di infiltrazione è l'infiltrazione del rivestimento interno della parete aortica (intima) e dei grandi vasi con colesterolo e suoi esteri, che è alla base di una malattia come l'aterosclerosi.
Decomposizione O fanerosi - questa è la rottura delle ultrastrutture cellulari e della sostanza intercellulare, che porta all'interruzione del metabolismo tissutale o cellulare e all'accumulo di prodotti del metabolismo compromesso in un tessuto o cellula, ad esempio, con intossicazione da difterite, si verifica la degenerazione grassa dei cardiomiociti. Sintesi perversa è la sintesi di sostanze nelle cellule e nei tessuti che non si trovano normalmente. Un esempio è l'amiloidosi, in particolare nel mieloma, quando i linfociti B, che normalmente producono anticorpi, producono proteine con struttura alterata durante la trasformazione del tumore.
Trasformazione – la formazione di prodotti di un tipo di scambio invece di prodotti di un altro tipo di scambio. Ad esempio, la trasformazione dei componenti di grassi e carboidrati in proteine.
Classificazione delle distrofie.
A seconda delle strutture in cui sono prevalentemente localizzati i cambiamenti distrofici, questi si dividono in parenchimali, stroma-vascolari (mesenchimali) e misti.
A seconda della predominanza dei disturbi dell'uno o dell'altro tipo di metabolismo, si distinguono le distrofie proteiche, grasse, carboidratiche, minerali e miste.
Si distinguono anche le distrofie acquisite ed ereditarie e, a seconda della prevalenza, generale e locale.
Distrofie parenchimali
In queste distrofie, le sostanze si accumulano nelle cellule del parenchima di vari organi, come miocardiociti, epatociti, neuroni, cellule dei tubuli renali e ghiandole surrenali. In base al tipo di disturbo metabolico, queste distrofie si dividono in proteiche (disproteinosi), grassi (lipidosi) e carboidrati.
Distrofie proteiche parenchimali (disproteinosi) caratterizzato da una violazione dello scambio di proteine citoplasmatiche in uno stato libero o legato . Questi includono la distrofia granulare, idropica, a goccia ialina e cornea.
Distrofia granulare .
Le cause della distrofia granulare possono essere disturbi circolatori, infezioni, intossicazioni e altri fattori che portano ad una diminuzione dell'intensità dei processi redox della cellula. In questo processo, gli organi risultano ingrossati, flaccidi, opachi e opachi quando vengono tagliati. Ciò dà motivo di chiamare la distrofia granulare un gonfiore torbido (opaco) degli organi.
Microscopicamente le cellule sono ingrandite, il loro citoplasma è torbido e ricco di granuli proteici. La microscopia elettronica nella distrofia granulare mostra un aumento del numero (iperplasia) e un rigonfiamento degli organelli cellulari, che otticamente assomigliano a granuli proteici. Tale iperplasia delle ultrastrutture cellulari è attualmente considerata come una pronunciata tensione funzionale degli organi a vari influssi.
L'esito della distrofia granulare è diverso. Nella maggior parte dei casi è reversibile, ma se non si eliminano le cause che l'hanno provocata, può trasformarsi in degenerazione gocciolina ialina, idropica o grassa.
La funzione degli organi colpiti può essere indebolita.
Distrofia delle goccioline ialine .
È un tipo più grave di distrofia. Molto spesso si sviluppa nei reni, nel fegato e meno spesso nel miocardio.
Con questo tipo di distrofia, nel citoplasma delle cellule compaiono grandi gocce di proteine, che spesso si fondono tra loro e, a seguito della coagulazione delle proteine, assomigliano alla sostanza fondamentale della cartilagine ialina.
L'aspetto degli organi solitamente non viene modificato o dipende dalle malattie in cui si verifica questa disproteinosi.
Nei reni, tale distrofia si sviluppa nei casi in cui una grande quantità di proteine penetra nelle urine attraverso il filtro renale. In questo caso, l'accumulo di inclusioni proteiche nel citoplasma e la sua distruzione sono causati dal mancato riassorbimento lisosomiale delle proteine dall'epitelio dei tubuli renali in condizioni di maggiore permeabilità del filtro glomerulare (nella sindrome nefrosica).
Nel fegato, la distrofia delle goccioline ialine si sviluppa spesso attraverso una sintesi perversa. Molto spesso si sviluppa con danno alcolico, quando nel citoplasma degli epatociti compaiono gocce di proteine ialine, chiamate corpi di Mallory (o ialine alcoliche) e sono un segno morfologico dell'epatite alcolica. A volte la degenerazione delle goccioline ialine degli epatociti può essere osservata nella cirrosi biliare e nella malattia di Wilson-Konovalov. L'ialina alcolica può avere un effetto citolitico sugli epatociti e stimolare la sintesi del collagene, che determina il decorso cronico progressivo dell'epatite alcolica e contribuisce allo sviluppo della cirrosi epatica.
La funzione organica nella distrofia delle goccioline ialine è solitamente compromessa in modo significativo. L'esito di questa distrofia può essere la necrosi coagulativa (morte) della cellula.
Distrofia idropica (idropisia).
Questa distrofia è caratterizzata dalla comparsa nelle cellule di vacuoli pieni di fluido citoplasmatico. Si verifica a causa di disturbi nel metabolismo dell'acqua, degli elettroliti e delle proteine, che si verificano durante intossicazioni di varia origine, nonché durante infezioni, soprattutto virali. Pertanto, si sviluppa nelle cellule dell'epidermide e delle mucose durante l'infezione erpetica, la varicella e il vaiolo naturale; negli epatociti – con epatite virale; nei neuroni del cervello e nei loro processi - nelle encefalopatie spongioformi (inclusa la malattia di Creutzfeldt-Jakob) causate da una molecola proteica infettiva - un prione. La degenerazione vacuolare può colpire i cilindri assiali delle fibre nervose del sistema di conduzione del midollo spinale nella leucospongiosi amiotrofica e le guaine mieliniche delle fibre nervose nell'infezione da HIV (mielopatia vacuolare).
L'aspetto degli organi di solito non viene modificato, ad eccezione della pelle durante l'infezione erpetica e il vaiolo, quando compaiono bolle (vescicole) piene di liquido sieroso.
All'esame microscopico, le cellule alterate risultano aumentate di volume, il loro citoplasma si riempie di vacuoli di varie dimensioni ed il nucleo è spostato verso la periferia della cellula. Man mano che il processo procede, i vacuoli si fondono tra loro e la cellula si trasforma in una vescicola, che consiste in un grande vacuolo e un nucleo vescicolare spostato ( distrofia del palloncino)
Nei reni, la distrofia idropica dell'epitelio del tubulo contorto può essere causata da un'insufficienza del sistema di riassorbimento del labirinto basale, che è troppo pieno di acqua che penetra nella cellula e, “risalindo” verso l'orletto a spazzola, distrugge le membrane e forma un palloncino strutture.
La degenerazione idropica degli epatociti è molto spesso il risultato dell'azione del virus dell'epatite B o di sostanze tossiche. Sotto l'influenza di un virus, questa disproteinosi avviene attraverso una sintesi perversa, subordinata alla riproduzione del virus, e sotto l'influenza di sostanze tossiche, è causata dall'insufficienza del sistema di disintossicazione.
Distrofia cornea
Questa condizione è caratterizzata dalla formazione eccessiva di sostanza corneo nell'epitelio squamoso cheratinizzante stratificato (pelle) o dalla cheratinizzazione dell'epitelio squamoso non cheratinizzante stratificato (esofago, cervice).
La distrofia cornea può manifestarsi con disturbi dello sviluppo cutaneo, disturbi circolatori cronici, infiammazioni croniche, infezioni, carenze vitaminiche, tumori, ecc.
La distrofia della pelle cornea può essere comune ( ittiosi- una malattia ereditaria che si manifesta con un'eccessiva cheratinizzazione - eccessivo desquamazione squamosa) e locale ( ipercheratosi).
La cheratinizzazione dell'epitelio squamoso stratificato non cheratinizzante, ad esempio nella cavità orale o nella cervice, appare macroscopicamente come una macchia bianca ( leucoplachia) .
Le disproteinosi parenchimali ereditarie, causate da una violazione del metabolismo intracellulare degli aminoacidi, comprendono: cistinosi, tirosinosi e oligofrenia fenilpiruvica (fenilchetonuria). Queste malattie colpiscono più spesso il fegato, i reni, il midollo osseo, la milza e il sistema nervoso.
Degenerazioni grasse parenchimali (lipidosi) ).
Queste distrofie sono caratterizzate da un alterato metabolismo dei grassi, principalmente neutri, nel citoplasma delle cellule. La predominanza dell'uno o dell'altro meccanismo per l'insorgenza della lipidosi dipende sia dalla causa che l'ha provocata sia dalle caratteristiche strutturali e funzionali dell'organo. Molto spesso, lo sviluppo della degenerazione grassa è associato all'ipossia, che si verifica con malattie croniche del sistema cardiovascolare, respiratorio, malattie del sistema sanguigno, nonché con l'azione di sostanze tossiche (varie infezioni e intossicazioni) e un aumento nel livello degli acidi grassi nel plasma sanguigno (obesità generale).
Tipicamente, la degenerazione grassa si sviluppa nel cuore, nel fegato e nei reni.
La degenerazione grassa del miocardio può verificarsi molto spesso a causa di ipossia o intossicazione (difterite, alcol, avvelenamento da fosforo, arsenico, ecc.). In entrambi i casi si sviluppano danni mitocondriali e una diminuzione dell'intensità dell'ossidazione degli acidi grassi.
Nelle cellule muscolari compaiono minuscole goccioline di grasso (obesità polverizzata) che, con cambiamenti crescenti (obesità da piccole goccioline), sostituiscono il citoplasma. Il processo è di natura focale e si osserva in gruppi di cellule muscolari lungo il ginocchio venoso dei capillari e nelle piccole vene, il che spiega l'aspetto peculiare del cuore: dall'endocardio sono visibili strisce trasversali gialle (cuore "tigre"). Il miocardio è flaccido, la dimensione del cuore può aumentare e le cavità si espandono.
L'esito della degenerazione grassa parenchimale del miocardio dipende dal grado della sua gravità. I cambiamenti iniziali sono reversibili, quelli profondi portano a gravi disfunzioni.
La degenerazione grassa del parenchima epatico è la più comune.
Può verificarsi quando il livello degli acidi grassi nel plasma sanguigno aumenta (diabete mellito, obesità generale), a seguito di decomposizione (con infezioni e intossicazioni), trasformazione (con alcolismo cronico), con malnutrizione per carenza di proteine in cibo, con difetti genetici degli enzimi.
Piccole goccioline di grasso compaiono nel citoplasma degli epatociti (obesità polverizzata), che si fondono in grandi goccioline (obesità a piccole goccioline). Di conseguenza, la gocciolina riempie quasi l'intero citoplasma, spingendo il nucleo verso la periferia della cellula (obesità da goccioline grandi). Macroscopicamente, il fegato si ingrandisce, diventa flaccido e acquisisce un colore giallo ocra o giallo-marrone (“fegato d’oca”).
La funzionalità epatica diminuisce drasticamente. Quando l'azione del fattore dannoso (ad esempio l'alcol, per più di 1 mese) cessa, la normale struttura della cellula può essere ripristinata. L'obesità su larga scala porta alla morte degli epatociti, a una reazione mesenchimale e allo sviluppo di fibrosi con conseguente cirrosi.
La degenerazione del rene grasso si sviluppa più spesso nelle cellule epiteliali dei tubuli prossimali e distali. per infiltrazione (con iperlipidemia), meno spesso - per decomposizione (con crescente ipossia). Molto spesso, i reni grassi si verificano nella sindrome nefrosica. I reni sono ingrossati, flaccidi, con macchie grigio-gialle sulla sezione. La funzione renale diminuisce.
Esiste un gruppo di lipidosi sistemiche ereditarie, che sono malattie da accumulo (thesaurismosi). Queste malattie sono associate a difetti ereditari degli enzimi che metabolizzano i grassi complessi. Questi includono: lipidosi cerebroside (malattia di Gaucher), lipidosi sfingomielinica (malattia di Niemann-Pick), lipidosi gangliosidica (malattia di Tay-Sachs o idiozia amaurotica precoce), gangliosidosi generalizzata (malattia di Norman-Landing), ecc. Il fegato, la milza, il midollo osseo e sistema nervoso centrale. La diagnosi morfologica è aiutata dalle cellule presenti nei tessuti caratteristici di un particolare tipo di lipidosi (cellule di Gaucher, cellule di Pick)
Distrofie parenchimali dei carboidrati. Caratterizzato da alterato metabolismo del glicogeno o delle glicoproteine.
Nel diabete mellito si sviluppano disturbi del metabolismo del glicogeno. In questa malattia si verifica una carenza assoluta e relativa di insulina, a seguito della quale l'utilizzo del glucosio e la sintesi del glicogeno vengono interrotti. Di conseguenza, il livello di glucosio nel sangue aumenta (iperglicemia), il glucosio appare nelle urine (glicosuria) e le riserve di glicogeno nel fegato e nei muscoli si esauriscono. In relazione all'iperglicemia e alla glicosuria, il glucosio si infiltra nell'epitelio tubulare dei reni e la sintesi del glicogeno avviene nell'epitelio tubulare, dove normalmente non esiste. Il citoplasma delle cellule dei tubuli renali diventa leggero e schiumoso.
Un eccessivo accumulo di glicogeno nelle cellule del fegato, dei reni e dei muscoli scheletrici si verifica anche nelle malattie ereditarie da accumulo (tesaurismosi) - glicogenosi causata da una carenza di vari enzimi che regolano il metabolismo del glicogeno (malattia di Gierke, Andersen, Pompe, McArdle, ecc.). )
Quando il metabolismo delle glicoproteine viene interrotto, mucine e mucoidi (sostanze simili al muco) si accumulano nelle cellule. Questo di solito si osserva durante i processi infiammatori nelle mucose, ad esempio con rinite, gastrite, bronchite, ecc. Tale muco può chiudere i lumi dei bronchi e dei dotti ghiandolari, il che porta alla formazione di cisti. A volte durante il gozzo colloidale vengono prodotte sostanze colloidali simili al muco nella ghiandola tiroidea. Il processo termina con l'atrofia delle mucose. Un eccessivo accumulo di muco si osserva anche nella fibrosi cistica (una malattia ereditaria sistemica). In questo caso, le cellule epiteliali delle ghiandole pancreatiche, dell'albero bronchiale, del tratto digerente e urinario, delle ghiandole sudoripare e lacrimali, dei dotti biliari) producono un muco viscoso e denso. I lumi delle ghiandole si ingrandiscono cisticamente e attorno a loro cresce il tessuto connettivo: fibrosi cistica.
DISTROFIE STROMALO-VASCOLARI (mesenchimali).
Le distrofie mesenchimali vengono rilevate nello stroma degli organi e nelle pareti dei vasi sanguigni e, a seconda del tipo di disturbo metabolico, sono suddivise in proteine, grassi e carboidrati.
A disproteinosi mesenchimale includono: gonfiore mucoide, gonfiore fibrinoide, ialinosi e amiloidosi.
A rigonfiamento mucoide nella sostanza principale del tessuto connettivo (di solito nelle pareti dei vasi sanguigni, nell'endocardio, nelle membrane sinoviali) si verifica l'accumulo e la ridistribuzione dei glicosaminoglicani, che hanno la proprietà di attrarre acqua, così come le proteine plasmatiche, principalmente le globuline.
Allo stesso tempo, le fibre di collagene si gonfiano e diventano non fibrose, ma rimangono preservate. L'accumulo di glicosaminoglicani presenta il fenomeno della metacromasia (la capacità di modificare il tono del colore di base), che consente facilmente di identificare i focolai di gonfiore mucoide nel tessuto connettivo. Questo fenomeno si manifesta più chiaramente quando colorato con blu di toluidina, quando i focolai di gonfiore mucoide non sono colorati di blu, ma lilla o rosso.
Questa distrofia si sviluppa più spesso nelle malattie infettive-allergiche (glomerulonefrite), allergiche (reazioni di ipersensibilità immediata) e autoimmuni (malattie reumatiche). L'aspetto dell'organo non viene modificato e il gonfiore del muco viene rilevato solo al microscopio. Quando la causa viene eliminata, può essere reversibile, altrimenti aumenta la disorganizzazione del tessuto connettivo e successivamente si sviluppa un gonfiore fibrinoide.
A gonfiore fibrinoide i tessuti continuano ad essere impregnati di proteine plasmatiche, che si accumulano nella sostanza fondamentale e nelle fibre di collagene, distruggendole e trasformandole in una massa omogenea contenente fibrinoide, una sostanza complessa costituita da fibrina, polisaccaridi, immunocomplessi (per i reumatismi), nucleoproteine (per lupus eritematoso sistemico). La metacromasia del tessuto connettivo non viene più rilevata, poiché si è verificata la distruzione dei glicosaminoglicani della sostanza principale. Il gonfiore fibrinoide è un processo irreversibile che può provocare necrosi fibrinoide, sclerosi o ialinosi.
A ialinosi nel tessuto connettivo si formano masse proteiche dense, traslucide, omogenee, che ricordano la cartilagine ialina (ialina).
La ialinosi può essere generale e locale. Si distinguono anche la ialinosi dei vasi sanguigni e il tessuto connettivo stesso.
La ialinosi può derivare da tre condizioni: gonfiore e necrosi dei fibrinoidi, sclerosi e impregnazione del plasma.
Ialinosi vascolare colpisce le piccole arterie e le arteriole e si verifica a seguito dell'impregnazione del plasma nell'ipertensione arteriosa e nel diabete mellito. A causa della deposizione delle proteine del plasma sanguigno nella parete, i vasi perdono la loro elasticità, il loro lume si restringe, con conseguente deterioramento dell'afflusso di sangue all'organo. La ialinosi vascolare è un processo sistemico ed è più pronunciato nei reni, nel cervello, nella retina, nel pancreas e nella pelle. La ialinosi delle arteriole cerebrali è particolarmente pericolosa, poiché improvvisi aumenti della pressione sanguigna (crisi ipertensive) possono portare alla rottura del vaso e all'emorragia nella sostanza del cervello.
Ialinosi del tessuto connettivo stesso si sviluppa a causa di gonfiore fibrinoide o sclerosi. Come risultato del rigonfiamento fibrinoide, la ialinosi si verifica in malattie associate a disturbi immunitari, come le malattie reumatiche, e di solito completa la progressiva distruzione del tessuto connettivo. Nei reumatismi, la ialinosi si verifica nelle valvole e nelle corde cardiache, il che contribuisce alla formazione di difetti cardiaci.
Di conseguenza sclerosi la ialinosi si forma nella pleura, negli strati pericardici e nel peritoneo dopo aver subito processi infiammatori. La ialinosi della capsula della milza porta alla formazione di una "milza vetrata": la capsula diventa spessa, satura di masse proteiche. La ialina può anche depositarsi nel tessuto connettivo della pelle dopo una lesione, soprattutto dopo ustioni al viso e al collo, provocando la formazione di cicatrici ruvide, dense e biancastre (cicatrici cheloidi).
Amiloidosi.
L'amiloidosi (latino amylum - amido) è una disproteinosi vascolare stromale, accompagnata da profondi cambiamenti nel metabolismo proteico e dalla comparsa di proteine fibrillari anomale - amiloide.
Nel 1884 Rokitansky chiamò l'amiloidosi una malattia sebacea, perché. gli organi colpiti hanno un aspetto untuoso. Successivamente, R. Virchow dimostrò che sotto l'influenza di iodio e acido solforico questa sostanza diventa blu e propose di chiamarla amiloide. La natura proteica dell'amiloide fu stabilita nel 1865 (M. Rudnev, Kuehne).
Microscopicamente, quando colorata con ematossilina ed eosina, l'amiloide appare come masse dense e eosinofile senza struttura. Per distinguere l'amiloide da altri depositi, vengono utilizzati metodi istochimici, ad esempio la colorazione con rosso Congo, iodio, viola di genziana e viola di metile, basati sul fenomeno della metacromasia, poiché le masse amiloidi contengono glicosaminoglicani.
In base alla sua struttura chimica si distinguono i seguenti tipi principali di amiloide:
Amiloide AA, che viene rilevata in alcuni tipi di amiloidosi ereditaria (febbre mediterranea familiare) e secondaria. Come risultato dell'attivazione del sistema dei fagociti monocitici nel fegato sotto l'influenza dell'interleuchina-1, viene stimolata la sintesi di SAA, che viene successivamente degradata con la formazione della proteina AA da cui si assemblano le fibrille amiloidi sulla superficie dei macrofagi ( amiloidoblasti);
Amiloide AL, rilevata nell'amiloidosi primaria e nella discrasia plasmacellulare neoplastica. In primo luogo, avviene la sintesi di catene leggere di immunoglobuline da cui vengono sintetizzate fibrille amiloidi da plasmacellule, macrofagi e cellule di mieloma;
L'amiloide FAP (AF) viene rilevata in alcuni tipi di amiloidosi ereditaria (polineuropatia amiloide familiare)
AS amiloide derivante dall'amiloidosi senile
La formazione di amiloide è strettamente correlata alle fibre del tessuto connettivo. L'amiloide può depositarsi lungo le fibre reticolari (amiloidosi perireticolare), ad esempio nella milza, nel fegato, nei reni, nell'intestino, e lungo le fibre di collagene (amiloidosi pericollagena), ad esempio nella muscolatura striata e liscia, nella pelle.
L'amiloidosi viene inoltre divisa in base alla deposizione predominante di amiloide negli organi in nefropatica, cardiopatica, neuropatica, epatica, ecc.
Si distinguono le seguenti forme cliniche e anatomiche di amiloidosi:
Amiloidosi idiopatica (primaria).. La causa e il meccanismo sono sconosciuti. In questo caso, l'amiloide appare nello stroma miocardico e nelle pareti vascolari (amiloidosi cardiopatica), nonché lungo i nervi periferici (amiloidosi neuropatica).
Amiloidosi ereditaria (genetica, familiare). Queste sono opzioni cardiopatiche, neuropatiche, meno spesso nefropatiche. Più comune nei paesi del Mediterraneo (Israele, Libano, ecc.)
Amiloidosi senile. L'amiloide si deposita nella corteccia cerebrale, essendo il centro delle placche senili (senili), così come nella parete dei piccoli vasi sanguigni. Tali cambiamenti compaiono in gran numero nella demenza senile (senile) e nel morbo di Alzheimer.
Amiloidosi acquisita (secondaria).. Si verifica più spesso ed è considerata una complicazione di varie malattie accompagnate da processi cronici suppurativi e distruttivi. L'amiloide si deposita sullo sfondo di qualsiasi malattia: tubercolosi; infiammazione cronica purulenta, ad esempio malattie polmonari non specifiche (bronchiectasie, polmonite cronica), osteomielite, ascessi cronici, sepsi cronica e malattie reumatiche (principalmente artrite reumatoide); e tumori del sangue (mieloma). L'amiloide si deposita solitamente nei reni, nella milza, nel fegato, nelle ghiandole surrenali e nell'intestino.
Gli organi aumentano di dimensioni, diventano densi e hanno un aspetto untuoso quando vengono tagliati.
Nella milza, l'amiloide si deposita prima nei follicoli linfatici sotto forma di grani traslucidi - milza di sago, quindi si accumula nella polpa rossa, la milza si ingrandisce, diventa densa, la superficie tagliata è liscia, lucida - milza sebacea.
L'amiloidosi renale è di grande importanza nella clinica, poiché è pericolosa per la vita del paziente. A partire dalle piramidi midollari renali, l’amiloidosi invade gradualmente la corteccia. L'amiloide si deposita in piccoli vasi, nel mesangio dei glomeruli, sulla membrana basale dei tubuli e nello stroma dell'organo.
Il bocciolo aumenta di dimensioni, la sua sostanza è densa, bianca, con una lucentezza untuosa (“grande bocciolo untuoso”). Emerge un quadro clinico e morfologico della nefrosi amiloide, che porta all'insufficienza renale cronica.
Anche il fegato affetto da amiloidosi è ingrossato, denso e appare “unto”.
Meno comunemente, l'amiloide si deposita nelle ghiandole surrenali, solitamente nella corteccia, e nell'intestino, nello strato sottomucoso.
Il significato funzionale è determinato dal grado di sviluppo dell'amiloidosi. L'amiloidosi grave porta alla degenerazione e all'atrofia del parenchima e alla sclerosi dello stroma dell'organo, portando al loro fallimento funzionale. Con l'amiloidosi grave, si osserva più spesso insufficienza renale cronica, meno spesso: insufficienza epatica, cardiaca, polmonare, surrenale, intestinale (sindrome da malassorbimento).
Definizione.Distrofia granulare- il risultato di un danno cellulare, caratterizzato dalla comparsa nel loro citoplasma di granuli, che sono mitocondri rigonfi. Distrofia idropica- rigonfiamento del citoplasma delle cellule dovuto all'eccessivo accumulo di acqua al suo interno. Nella maggior parte dei casi accompagna la distrofia granulare e presenta meccanismi comuni con essa.
Evento. Entrambe le condizioni sono più spesso osservate negli epatociti, nei nefrociti e nei cardiomiociti. Il fenomeno è estremamente comune, poiché si manifesta principalmente durante l'ipossia circolatoria, che si verifica in molte condizioni patologiche.
Condizioni di accadimento. Un prerequisito per il verificarsi di questi tipi di danni è l'ipossia, che può essere causata da:
1) un calo della pressione sanguigna sistemica o un'interruzione locale del flusso sanguigno arterioso, accompagnato da ipoperfusione tissutale;
2) relativa insufficienza (inadeguatezza) dell'apporto di sangue all'organo in condizioni di intenso funzionamento;
3) ipossia tissutale in condizioni di edema tissutale, manifestata da una ridotta diffusione dell'ossigeno dai capillari alle cellule;
4) ipossia ipossica associata a bassa saturazione di ossigeno nel sangue;
5) ipossia ematica nell'anemia, manifestata da una carenza nel sangue o da un'inferiorità dei trasportatori di ossigeno - eritrociti e/o emoglobina. Una variante dell'ipossia emica può essere la ridotta dissociazione dell'ossiemoglobina e il ridotto rilascio di ossigeno da parte degli eritrociti in determinate condizioni patologiche.
Meccanismi di accadimento. In condizioni ipossiche nella cellula, l'intensità della fosforilazione ossidativa e della sintesi di ATP diminuisce drasticamente. A causa della carenza di energia, il lavoro delle pompe ioniche - K + /Na + -ATPasi - integrate nelle membrane degli organelli e delle cellule e che garantiscono la rimozione attiva degli ioni Na + all'esterno della cellula, viene interrotto. Questi ioni e, di conseguenza, l'acqua si accumulano negli organelli. Ciò è più evidente nei mitocondri, che di conseguenza si gonfiano e, se osservati al microscopio ottico, sembrano grani nel citoplasma di una cellula precedentemente omogenea (Fig. 34.1).
Il rigonfiamento dei mitocondri e la frammentazione delle creste al loro interno interrompono ulteriormente la sintesi di ATP. Anche la "danza dei mitocondri" normalmente esistente viene bruscamente interrotta: il loro movimento attraverso il citoplasma, grazie al quale l'energia viene consegnata ad altri organelli: i mitocondri che sono diventati "goffi" assicurano scarsamente la consegna di ATP, a causa della quale il lavoro di altri gli organelli soffrono.
Poiché gli ioni Na + e l'acqua si accumulano non solo negli organelli, ma anche nel citoplasma della cellula, si gonfia e l'accumulo di acqua nelle cisterne del reticolo endoplasmatico ruvido, che avviene con lo stesso meccanismo, rende il citoplasma cellulare schiumoso se osservato al microscopio ottico. Il grado estremo di distrofia idropica è designato come distrofia del palloncino.
Tutto ciò si sviluppa abbastanza rapidamente, ad esempio, si osserva una degenerazione granulare degli epatociti già in caso di morte dopo 5 minuti per grave trauma, accompagnata da un calo della pressione sanguigna e da ipoperfusione epatica.
Questo fenomeno è reversibile. Se l'ipossia si interrompe, la sintesi di ATP riprende, le pompe ioniche rimuovono il Na + in eccesso dal citoplasma e dagli organelli, dopodiché l'acqua in eccesso li lascia. I mitocondri ripristinano le loro dimensioni e struttura interna, e quelli che hanno perso lo strato esterno della membrana vengono catturati dai lisosomi e utilizzati in essi. Se l'ipossia continua, si sviluppa un danno irreversibile agli organelli, che porta alla morte cellulare.
Immagine macroscopica. I cambiamenti negli organi interni in questo tipo di distrofia granulare a livello macroscopico sono descritti come gonfiore nuvoloso. Nei reni e nel fegato, che hanno una capsula, il tessuto si gonfia leggermente oltre i bordi del taglio, anche se questo non è sempre pronunciato. Il rigonfiamento è associato all'accumulo di quantità eccessive di acqua nelle cellule, già descritto sopra. La lucentezza opaca e l'aspetto opaco del tessuto sulla sezione sono spiegati dal fatto che lo strato superficiale della sezione risulta essere otticamente più denso a causa dei mitocondri gonfiati, per cui il tessuto trasmette meno bene e riflette maggiormente la luce incidente .
Immagine microscopica. Nel citoplasma delle cellule, ad alto ingrandimento, si rivelano piccoli grani che assumono un colore rosa intenso quando colorati con eosina (Fig. 34.2). Sono particolarmente evidenti quando il preparato non viene colorato con ematossilina ed eosina, ma con azzurro ed eosina.
A livello ultrastrutturale, i mitocondri sono ingranditi di 2-5 volte o più, localizzati in prossimità di una matrice ripulita, con creste frammentate, spesso scarsamente visibili (Fig. 34.3). Lo strato esterno della membrana a doppio strato è assente in alcuni dei mitocondri più modificati. Il cambiamento nei mitocondri è accompagnato da un'espansione più o meno pronunciata del reticolo endoplasmatico.
La distrofia idropica a livello luce-ottico si manifesta con la schiumosità del citoplasma (Fig. 34.4). Molto spesso si osserva nell'epitelio dei tubuli renali. La microscopia elettronica rivela cisterne dilatate del reticolo endoplasmatico e del complesso di Golgi (Fig. 34.5a).
Significato clinico. Il fatto stesso della distrofia granulare indica una carenza di ossigeno ed energia nella cellula, che ha un effetto estremamente sfavorevole sulla sua funzione, soprattutto quando si tratta di cardiomiociti. Ciò non sorprende: in condizioni di carenza energetica, la cellula “si preoccupa” non degli interessi del corpo, ma della propria integrità, quindi non ha tempo per contrarsi, pinocitosi o sintesi di nulla - la maggior parte dei pochi ATP Le molecole formate durante la glicolisi anaerobica vanno a garantire il lavoro delle pompe ioniche.
Anche il rigonfiamento cellulare è considerato un fattore che può compromettere la circolazione sanguigna nei capillari adiacenti.
Informazioni correlate.
Tradotto letteralmente dal greco, distrofia significa “disturbi alimentari”. Questo termine combina cambiamenti quantitativi e qualitativi nelle strutture dei tessuti e negli organi, che, a loro volta, portano ai loro disturbi funzionali. Le deviazioni distrofiche possono essere generali e locali e hanno un secondo nome: nefrosi, ma viene utilizzato in rari casi.
Tra le persone, si sente parlare più spesso della distrofia renale granulare, poiché è proprio questa che è associata a un disturbo del metabolismo proteico, che si osserva in molti pazienti. Ed è anche quello che più spesso coinvolge cuore e fegato nel processo patologico. La classificazione di tali disturbi è molto ampia. Convenzionalmente possono essere suddivisi nelle seguenti categorie:
- cambiamenti associati al metabolismo;
- Locale;
- generalizzato;
- congenito e acquisito;
- mesenchimale, parenchimale e misto.
caratteristiche generali
I disturbi distrofici nei reni hanno una localizzazione chiara, cioè si trovano all'interno di un organo. Ma nonostante ciò, il sistema urinario inizia successivamente a soffrire, e quindi l'intero corpo nel suo insieme.

Per fare una diagnosi accurata è necessario sottoporsi a una serie di procedure diagnostiche, poiché ogni tipo di distrofia ha le sue caratteristiche
I reni svolgono una funzione escretoria. Sono necessari affinché una persona possa liberare il corpo dai prodotti di decomposizione che si formano a seguito del metabolismo. I processi metabolici si verificano nei glomeruli dei reni, dove avviene l'interazione tra sangue e soluzione acquosa, ed è in essi che si accumulano tutti i componenti non necessari per il corpo umano. Tutte le sostanze tossiche vengono rilasciate anche attraverso i reni, quindi l'organo accoppiato soffre sempre di vari avvelenamenti, nella stessa misura del fegato.
Distrofie associate a disturbi del metabolismo proteico
A causa del fatto che i reni partecipano attivamente al metabolismo delle proteine, ai pazienti viene spesso diagnosticata una distrofia proteica. Nel processo sono coinvolte le strutture cellulari del parenchima; ciò avviene a causa di formazioni purulente, patologie del tessuto connettivo, neoplasie maligne e infiammazioni croniche.
Il gonfiore dei tessuti è il segno principale del processo patologico; inoltre, quando si esegue un test generale delle urine, viene rilevato un gran numero di globuli rossi, leucociti e composti proteici. Ma questo metodo strumentale non è sufficientemente informativo, poiché un tale quadro clinico accompagna la maggior parte delle malattie del sistema urinario.
Le distrofie proteiche includono:
- granuloso;
- idropico;
- goccia ialina.
Distrofia granulare
La distrofia granulare dell'epitelio dei tubuli contorti del rene ha un nome così dettagliato, poiché all'esame istologico si può notare che la forma di queste strutture cambia. La malattia può colpire il fegato e il cuore e può essere definita gonfiore opaco o opaco. La ragione principale per lo sviluppo del processo patologico è considerata la presenza di infezione o avvelenamento acuto.

Se i segni di avvelenamento vengono eliminati molto rapidamente, il rene tornerà al suo stato originale
Con una diagnosi tempestiva e l'eliminazione dei segni di intossicazione, il rene tornerà rapidamente al suo stato precedente. Ma se il processo di penetrazione delle sostanze tossiche in profondità nel corpo non viene rallentato, inizierà lo stadio di necrosi dell'organo accoppiato. Il secondo organo, dopo il rene, coinvolto nel processo di distrofia è il fegato. Perde il colore, sbiadisce, o meglio, assume una sfumatura di marciume, mentre la granulosità sul fegato può essere assente o del tutto assente.
Per quanto riguarda il cuore, aumenta di dimensioni e il tessuto muscolare diventa flaccido. Non sono presenti focolai granulari, ma si osservano strutture cellulari basofile e ossofile. Il muscolo cardiaco diventa più sensibile ai coloranti blu e lilla.
Distrofia idropica
La distrofia idropica dell'epitelio tubulare ha anche un secondo nome, vale a dire distrofia vacuolare. Ciò è dovuto al fatto che nelle strutture cellulari si formano grandi vacuoli contenenti fluido intracellulare.
Le ragioni che portano allo sviluppo di questa patologia includono: diabete mellito, amiloidosi, forte diminuzione del livello di potassio nel sangue, glomerulonefrite, intossicazione da glicole. Tutte le malattie sopra menzionate portano ad un'accelerazione del processo di filtrazione, con conseguente interruzione delle funzioni enzimatiche. Sfortunatamente, la prognosi per il recupero non è delle più favorevoli, poiché molto spesso ciò porta allo sviluppo di aree di necrosi renale.
Distrofia delle goccioline ialine
La distrofia delle goccioline ialine colpisce le aree dell'epitelio che riveste i tubuli contorti dei reni. Un certo numero di cellule epiteliali partecipano ai processi metabolici che avvengono tra la matrice renale e il sangue.
La distrofia delle goccioline dell'epitelio si sviluppa a causa della presenza nel corpo di malattie come la nefrite glomerulare, la pielonefrite e le intossicazioni di vario tipo. Disturbi di questa natura sono così chiamati perché nelle strutture cellulari si accumulano goccioline di una sostanza simile alla ialina; sono un elemento diagnostico.
Distrofia cornea
La distrofia cornea è associata all'accumulo eccessivo di una sostanza specifica: la cheratina. Questo composto proteico si trova nelle cellule epiteliali. Se la cellula è costantemente esposta a un fattore dannoso come le fibrille citoplasmatiche, la quantità di cheratina aumenterà.
Cambiamenti distrofici associati a disturbi del metabolismo lipidico
La degenerazione dei reni grassi ha un secondo nome: lipoide. In una persona completamente sana è normale la presenza di un piccolo numero di cellule adipose nei reni, che si trovano nella zona dei dotti collettori. Ma nei pazienti con grave obesità, i depositi lipidici iniziano ad accumularsi nei tubuli prossimali e distali dei reni. I depositi di grasso si dividono in colesterolo, fosfolipidi e lipidi liberi.

La degenerazione lipoide si osserva più spesso nei pazienti in sovrappeso
Gli organi accoppiati iniziano ad aumentare di dimensioni e iniziano anche a ricoprirsi di uno strato grasso, colorato di giallo. I reni iniziano a ricoprirsi di lipidi a causa della degenerazione dei tessuti o della carenza di ossigeno (patologie dell'apparato respiratorio, insufficienza cardiaca, anemia, abuso di alcol). Inoltre, le ragioni per lo sviluppo di questa malattia includono:
- intossicazione da cloroformio;
- fosforo o arsenico;
- la presenza di un processo infettivo;
- dieta a basso contenuto proteico;
- avitaminosi.
Distrofia del glicogeno
Questo tipo di disturbo distrofico si osserva nei pazienti con diabete. Caratterizzato da una quantità eccessiva di glicogeno nel nefrone distale e nelle anse. Il glicogeno è necessario per lo stoccaggio e il successivo accumulo di composti di carboidrati nel corpo umano.
Tipi specifici di nefrosi
Esistono due tipi di disturbi distrofici e il termine nefrosi viene spesso applicato ad essi: nefrosi febbrile e necrotizzante.
Nefrosi febbrile
La nefrosi febbrile si sviluppa a causa della rapida patologia infettiva nel corpo umano. Per determinare questo tipo di distrofia, è necessario superare un esame generale delle urine, in cui verranno rilevate le cellule epiteliali renali. Se si cura una malattia infettiva, anche la nefrosi scomparirà, cioè non saranno necessari farmaci specifici.
Nefrosi necrotizzante
La causa dello sviluppo della nefrosi necrotica è l'intossicazione acuta del corpo con veleni renali. Questi includono: acidi di origine organica, mercurio, piombo, cromo. Inoltre, la necrosi del tessuto renale può essere causata da malattie infettive e danni a vaste aree della pelle (ustioni).
Misure diagnostiche
Il danno renale distrofico potrebbe non manifestarsi per un periodo piuttosto lungo, cioè potrebbe essere asintomatico. L'alba del quadro clinico cade nel periodo in cui l'organo accoppiato cessa di far fronte alle sue responsabilità funzionali. È in questo momento che le sostanze assenti nelle persone sane iniziano ad accumularsi nel corpo umano.
L’obiettivo principale delle procedure diagnostiche è determinare la presenza di questi componenti nelle urine e nel sangue della vittima.
Ogni tipo specifico di distrofia ha determinati prodotti metabolici ed è da essi che si può determinare il tipo di processo patologico. Ad esempio, se le strutture cellulari dell'epitelio renale vengono determinate nelle urine del paziente, è possibile giudicare la nefrosi febbrile e, se è presente la proteina amiloide, viene diagnosticata la nefrosi amiloide.

La diagnosi tempestiva consentirà di individuare la malattia nelle fasi iniziali
Per una diagnosi più accurata, vengono utilizzati metodi ad ultrasuoni. Quando si esegue tale manipolazione, è possibile notare un aumento delle dimensioni del rene e, nella sua struttura, diventa flaccido e sciolto. Qualsiasi processo patologico che si verifica nel corpo influisce negativamente sulla condizione dei reni. Inoltre, si osserva un particolare effetto negativo durante l'assunzione di vari farmaci, che allo stesso tempo sono vitali.
L'organo accoppiato è molto sensibile a varie anomalie, quindi vale la pena monitorare le sue condizioni e, al minimo problema o segno di malattia, consultare un medico. Vale la pena ricordare che la maggior parte delle lesioni renali distrofiche nelle fasi iniziali sono facilmente curabili e sono un processo reversibile. Se la nefrosi non viene trattata, può portare alla completa perdita del rene.
La distrofia è un processo patologico complesso, che si basa su una violazione del metabolismo dei tessuti che porta a cambiamenti strutturali.
I trofici sono un insieme di meccanismi. determinare il metabolismo e l'organizzazione strutturale della cellula (tessuto) necessari per svolgere una funzione specializzata.
Cause delle distrofie:
1) disturbi dell'autoregolazione cellulare, che possono essere causati da iperfunzione, sostanze tossiche, radiazioni, carenza di enzimi, ecc.
2) la disfunzione dei sistemi di trasporto che assicurano il metabolismo e la conservazione strutturale dei tessuti provoca ipossia.
3) interruzione della regolazione endocrina e nervosa
Morfogenesi delle distrofie:
1) infiltrazione
Accumulo eccessivo di una sostanza (normale, non anormale) a causa di un'eccessiva sintesi.
Esempio: epatosi del fegato grasso, emosiderosi renale.
2) decomposizione (fanerosi)
Disintegrazione delle ultrastrutture cellulari e della sostanza intercellulare, con conseguente interruzione del metabolismo dei tessuti e accumulo di prodotti del metabolismo compromesso nel tessuto.
3) sintesi perversa
Sintesi di prodotti anomali. Questi includono: sintesi della proteina amiloide anormale nella cellula, sintesi della proteina ialina alcolica da parte degli epatociti.
4) trasformazione
La formazione di prodotti di un tipo di scambio da prodotti iniziali comuni che entrano nella costruzione di BZHU.
Classificazioni della distrofia.
La classificazione aderisce a diversi principi. Le distrofie si distinguono:
1) per dominanza cambiamenti morfologici nelle strutture tissutali: parenchimale, misto, mesenchimale (stroma-vascolare)
2) per dominanza violazioni dell'uno o dell'altro tipo di scambio. proteine, grassi, carboidrati, minerali.
3) a seconda influenza di fattori genetici. acquisito, ereditario.
4) per localizzazione. locale, generale.
Distrofie parenchimali.
Manifestazioni di disordini metabolici in cellule funzionalmente altamente specializzate.
1) Distrofie proteiche parenchimali (disproteinosi)
L'essenza di tali distrofie è un cambiamento nelle proprietà fisico-chimiche e morfologiche delle proteine cellulari: subiscono denaturazione e coagulazione o colliquazione, che porta all'idratazione del citoplasma. Nei casi in cui i legami tra proteine e lipidi vengono interrotti, si verifica la distruzione delle strutture della membrana cellulare.
I disturbi del metabolismo proteico sono spesso associati a disturbi della pompa Na-K: ciò porta all'accumulo di ioni Na e al rigonfiamento cellulare. Questo processo patologico è chiamato distrofia idropica.
-granuloso
Reversibile, assomiglia all'accumulo di piccoli granelli proteici nel citoplasma. Gli organi aumentano di dimensioni, diventano flaccidi e opachi.
-flebo ialino
Nel citoplasma compaiono grandi goccioline proteiche di tipo ialino, che si fondono tra loro e riempiono il corpo cellulare. In alcuni casi termina con una necrosi coagulativa focale della cellula.
Spesso si trova nei reni, raramente nel fegato e nel miocardio.
Nei reni, quando esaminati, si riscontra un accumulo di goccioline nei nefrociti. L'accumulo è spesso osservato nella sindrome nefrosica, poiché la base di questa distrofia è l'insufficienza dell'apparato vacuolare-lisosomiale dell'epitelio del tubulo prossimale, in cui le proteine vengono normalmente riassorbite. Questo è il motivo per cui nelle urine compaiono proteine (proteinuria) e cilindri (cilindruria).
L'aspetto non ha caratteristiche caratteristiche.
Nel fegato, la microscopia rivela corpi Malory, costituiti da fibrille e ialine alcoliche. L'aspetto di tali gocce è una manifestazione della funzione sintetica perversa dell'epatocita, che si verifica nell'epatite alcolica e nella cirrosi biliare primaria. L'aspetto del fegato varia.
L'esito della distrofia delle goccioline ialine è sfavorevole e porta alla necrosi cellulare.
-distrofia idropica
Caratterizzato dalla comparsa nella cellula di vacuoli pieni di fluido citoplasmatico. Si osserva più spesso nell'epitelio della pelle e nei tubuli renali, negli epatociti e nei miociti.
Le cellule parenchimali aumentano di volume, il loro citoplasma si riempie di vacuoli contenenti liquido limpido. Quindi la cellula si trasforma in un enorme palloncino (l'intera cellula è diventata un grande vacuolo) - necrosi focale da liquefazione. L'aspetto dei tessuti cambia poco.
Un ruolo importante nel meccanismo di sviluppo è giocato dalla ridotta permeabilità della membrana, che porta all'acidificazione del citoplasma e all'attivazione degli enzimi idrolitici dei lisosomi, che rompono i legami intramolecolari con l'aggiunta di acqua.
Cause: nei reni - danno al filtro renale, che porta all'iperfiltrazione, nel fegato - epatite di varie eziologie, nell'epidermide - edema, infezione.
L'esito di tale distrofia è solitamente sfavorevole: termina con una necrosi coagulativa focale.
-distrofia cornea
È caratterizzata da un'eccessiva formazione di sostanza corneo nell'epitelio cheratinizzante (ipercheratosi, ittiosi) o dalla formazione di sostanza cornea dove normalmente non esiste (cheratinizzazione patologica delle mucose). Le ragioni sono molteplici: disturbi dello sviluppo cutaneo, infiammazioni croniche, carenze vitaminiche, ecc.
Risultato: a volte, quando la causa viene eliminata, si verifica il ripristino dei tessuti, ma nei casi avanzati si verifica la morte cellulare.
- disturbi ereditari del metabolismo degli aminoacidi
Le cosiddette malattie da accumulo, che si basano su una violazione del metabolismo intracellulare di un numero di aminoacidi a causa della carenza ereditaria di enzimi metabolizzanti.
A) cistinosi. La scienza non sa ancora quale carenza di enzima porta a questa malattia. Gli AA si accumulano nel fegato, nei reni, nella milza, negli occhi, nel midollo osseo e nella pelle.
B) tirosinosi. Si verifica a causa del deficit di tirosina aminotransferasi. Si accumula nel fegato, nei reni, nelle ossa.
B) oligofrenia fenilpiruvica. Si verifica quando c'è una carenza di fenilalanina 4-idrossilasi e si accumula nel sistema nervoso, nei muscoli e nel sangue.
2) Degenerazioni grasse parenchimali (lipidosi)
I disturbi nel metabolismo dei lipidi citoplasmatici possono manifestarsi con un aumento del loro contenuto nelle cellule, dove si trovano normalmente. nell'aspetto dei lipidi dove solitamente non si trovano e nella formazione di grassi di composizione chimica insolita.
- disturbi del metabolismo lipidico
Nel fegato, la degenerazione grassa si manifesta con un forte aumento del contenuto di grassi negli epatociti e un cambiamento nella loro composizione. Nelle cellule epatiche compaiono prima i granuli lipidici (obesità polverizzata), poi piccole goccioline (obesità da piccole goccioline), che poi si fondono in grandi goccioline (obesità da goccioline grandi) o in un vacuolo di grasso. Il fegato è ingrossato, flaccido e di colore giallo ocra. Tra i meccanismi di degenerazione del fegato grasso ricordiamo un eccessivo apporto di acidi grassi negli epatociti o la loro aumentata sintesi da parte di queste cellule, l'esposizione a sostanze tossiche che bloccano l'ossidazione degli acidi grassi e la sintesi delle lipoproteine negli epatociti e un insufficiente apporto di aminoacidi acidi necessari per la sintesi nelle cellule del fegato. Pertanto, l'HDP si verifica a seguito di: lipoproteinemia (alcolismo, diabete mellito, obesità generale), intossicazioni epatotrope (etanolo, cloroformio) e disturbi nutrizionali.
La degenerazione grassa del miocardio si verifica a causa dell'ipossia e dell'intossicazione. Il meccanismo di sviluppo è associato ad una diminuzione dell'ossidazione degli acidi grassi dovuta alla distruzione dei mitocondri sotto l'influenza dell'ipossia o della tossina. All'esame macroscopico la dimensione del cuore risulta aumentata, il muscolo cardiaco è di colore giallo argilla. Il miocardio assomiglia alla pelle di una tigre: striature bianche e gialle. I lipidi sono determinati sotto forma di piccole gocce.
Le cause della degenerazione grassa sono varie. Possono essere associati alla carenza di ossigeno (motivo per cui si riscontra spesso nelle malattie del sistema cardiovascolare), infezioni e intossicazioni, carenze vitaminiche e alimentazione unilaterale.
L'esito della degenerazione grassa dipende dal suo grado. Se non è accompagnato da una grave disgregazione delle strutture cellulari, è reversibile.
-enzimopatie ereditarie
Sorgono a causa della carenza ereditaria di enzimi coinvolti nel metabolismo dei lipidi.
A) Malattia di Gaucher da deficit di glucocerebrosidasi. I lipidi si accumulano nel fegato, nella milza e nel midollo osseo.
B) Malattia di Niemann-Pick da deficit di sfingomielinasi. Accumulo nel fegato, nella milza, nel midollo osseo.
B) Malattia di Sachs da deficit di galattosidasi acida.
D) Malattia di Norman-Landing da deficit di beta-galattosidasi.
3) Distrofie parenchimali dei carboidrati
-distrofie dei carboidrati associate ad alterato metabolismo del glicogeno
Nel diabete mellito vi è un uso insufficiente del glucosio da parte dei tessuti, un aumento del suo contenuto nel sangue e l'escrezione nelle urine. Le riserve di glicogeno tissutale diminuiscono drasticamente. Nel fegato, la sintesi del glicogeno viene interrotta, il che porta alla sua infiltrazione di grassi e alla degenerazione del fegato grasso.
Nei reni affetti da diabete mellito si verificano i seguenti cambiamenti: infiltrazione di glicogeno nell'epitelio tubulare.
- glicogenosi ereditaria
a) tipo 1 – malattia di Gierke – deficit di glucosio-6-fosfatasi
b) tipo 2 – Malattia di Pompe – deficit di alfa-1,4-glucosidasi acida
c) tipo 3 – malattia di Forbes – deficit di amilo-1,6-glucosidasi
d) tipo 4 – malattia di Anderson – deficit di amilo-(1,4-1,6)-transglucosidasi
e) tipo 5 – malattia di McArdle – deficit di miofosforilasi
e) tipo 6 – Malattia di Hers – deficit di fosforilasi epatica
Nelle malattie di tipo 1,2,5,6, la struttura del glicogeno non è disturbata.
-distrofie dei carboidrati associate a disturbi del metabolismo delle glicoproteine
Nelle cellule o nella sostanza intercellulare si verifica l'accumulo di mucine e mucoidi, chiamate anche sostanze mucose o simili al muco.
Molte cellule secernenti muoiono e desquamano, i dotti escretori delle ghiandole vengono ostruiti dal muco, il che porta allo sviluppo di cisti.
Le cause sono varie, ma molto spesso si tratta di infiammazione delle mucose a causa dell'azione di vari agenti patogeni irritanti.
Maggiori informazioni
Distrofia granulare del rene (microimmagine) Distrofia granulare (gonfiore torbido) del rene. Microimmagine. Colorazione G-E. (secondo V. A. Salimov) A (grandezza 240): 1. nuclei delle cellule epiteliali tubulari renali; 2. glomerulo vascolare; 3. lotto contorto; 4. accumulo di proteine (cast proteici) nel lume del tubulo contorto.
 Distrofia granulare del rene (microimmagine) Distrofia granulare (gonfiore torbido) del rene. Microimmagine. Colorazione G-E. (secondo V. A. Salimov) B (punteggio 960): 1. epitelio dei tubuli contorti (si nota la granularità fine, simile a una polvere del citoplasma, le cellule sono gonfie, non ci sono nuclei, i confini tra le cellule non sono visibili); 2. cilindro proteico; 3. glomerulo renale (vascolare).
Distrofia granulare del rene (microimmagine) Distrofia granulare (gonfiore torbido) del rene. Microimmagine. Colorazione G-E. (secondo V. A. Salimov) B (punteggio 960): 1. epitelio dei tubuli contorti (si nota la granularità fine, simile a una polvere del citoplasma, le cellule sono gonfie, non ci sono nuclei, i confini tra le cellule non sono visibili); 2. cilindro proteico; 3. glomerulo renale (vascolare).
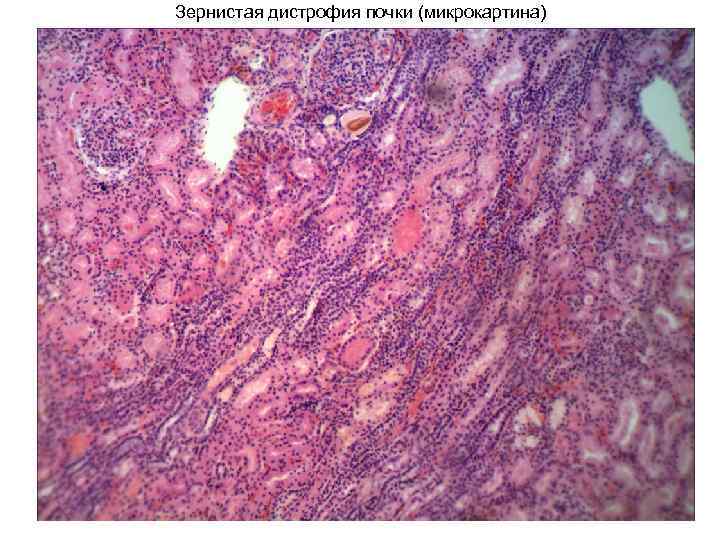

 Distrofia granulare del rene (immagine macro) Distrofia granulare (gonfiore torbido) del rene. Immagine macro. (secondo V. A. Salimov) 1. strato corticale; 2. confine levigato tra gli strati corticale e midollo del rene; 3. midollo renale; 4. gemmazione dalla superficie; 5. emorragie renali
Distrofia granulare del rene (immagine macro) Distrofia granulare (gonfiore torbido) del rene. Immagine macro. (secondo V. A. Salimov) 1. strato corticale; 2. confine levigato tra gli strati corticale e midollo del rene; 3. midollo renale; 4. gemmazione dalla superficie; 5. emorragie renali
 Distrofia granulare del rene (microimmagine) 1. Tubulo renale senza cambiamenti visibili; 2. Tubuli renali in uno stato di distrofia granulare: A) accumulo di granelli simili a polvere di natura proteica nelle cellule epiteliali dei tubuli renali, B) lume bizzarramente alterato del tubulo renale
Distrofia granulare del rene (microimmagine) 1. Tubulo renale senza cambiamenti visibili; 2. Tubuli renali in uno stato di distrofia granulare: A) accumulo di granelli simili a polvere di natura proteica nelle cellule epiteliali dei tubuli renali, B) lume bizzarramente alterato del tubulo renale


 Distrofia epatica granulare 1. Epatociti in uno stato di distrofia granulare e necrobiosi; 2. Citoplasma degli epatociti, aumentato di volume, eterogeneo, con granulosità fine e limpida; 3. Epatociti che hanno perso il nucleo (in stato di necrobiosi)
Distrofia epatica granulare 1. Epatociti in uno stato di distrofia granulare e necrobiosi; 2. Citoplasma degli epatociti, aumentato di volume, eterogeneo, con granulosità fine e limpida; 3. Epatociti che hanno perso il nucleo (in stato di necrobiosi)

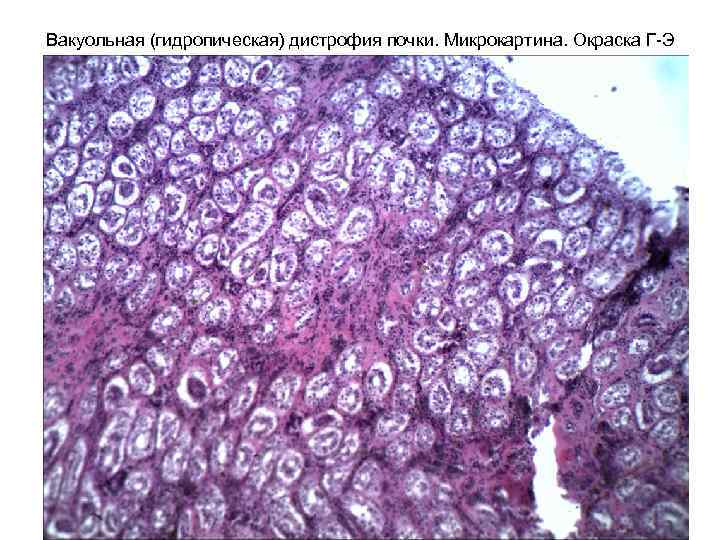


 Distrofia renale vacuolare (idropica). Microimmagine. Colorazione G-E 1. Presenza di palloncini e vacuoli nel citoplasma delle cellule epiteliali renali; 2. Disintegrazione dei nuclei e del citoplasma a seguito della loro compressione da parte dei vacuoli (formazione di “cellule ad anello”)
Distrofia renale vacuolare (idropica). Microimmagine. Colorazione G-E 1. Presenza di palloncini e vacuoli nel citoplasma delle cellule epiteliali renali; 2. Disintegrazione dei nuclei e del citoplasma a seguito della loro compressione da parte dei vacuoli (formazione di “cellule ad anello”)


 Distrofia delle goccioline ialine del rene. Microimmagine della distrofia renale delle goccioline ialine. Microimmagine. Colorazione G-E. (secondo V.V. Serov, N.E. Yarygin, V.S. Paukov) a. il citoplasma delle cellule epiteliali dei tubuli renali è pieno di grandi goccioline di natura proteica
Distrofia delle goccioline ialine del rene. Microimmagine della distrofia renale delle goccioline ialine. Microimmagine. Colorazione G-E. (secondo V.V. Serov, N.E. Yarygin, V.S. Paukov) a. il citoplasma delle cellule epiteliali dei tubuli renali è pieno di grandi goccioline di natura proteica
 Distrofia delle goccioline ialine del rene. Microimmagine 1. Accumulo di gocce ialine nel citoplasma delle cellule epiteliali del tubulo renale (tale accumulo provoca atrofia e disintegrazione del citoplasma e del nucleo); 2. rilascio di gocce ialine nel lume dei tubuli renali
Distrofia delle goccioline ialine del rene. Microimmagine 1. Accumulo di gocce ialine nel citoplasma delle cellule epiteliali del tubulo renale (tale accumulo provoca atrofia e disintegrazione del citoplasma e del nucleo); 2. rilascio di gocce ialine nel lume dei tubuli renali



 Ialinosi dei vasi uterini 1. Vaso sanguigno in stato di ialinosi; 2. Una parete della nave fortemente ispessita a causa della deposizione di ialino in essa; 3. “Accoppiamento tessuto”; 4. Lume a fessura del vaso fortemente ristretto; 5. Ghiandole uterine conservate
Ialinosi dei vasi uterini 1. Vaso sanguigno in stato di ialinosi; 2. Una parete della nave fortemente ispessita a causa della deposizione di ialino in essa; 3. “Accoppiamento tessuto”; 4. Lume a fessura del vaso fortemente ristretto; 5. Ghiandole uterine conservate


 Ialinosi della capsula e dello stroma della milza 1. Depositi di ialino nella capsula della milza (la capsula assume un aspetto omogeneo e aumenta notevolmente di volume); 2. Sezioni longitudinali e trasversali delle trabecole della milza, sature di ialina; 3. Il parenchima della milza è invariato.
Ialinosi della capsula e dello stroma della milza 1. Depositi di ialino nella capsula della milza (la capsula assume un aspetto omogeneo e aumenta notevolmente di volume); 2. Sezioni longitudinali e trasversali delle trabecole della milza, sature di ialina; 3. Il parenchima della milza è invariato.
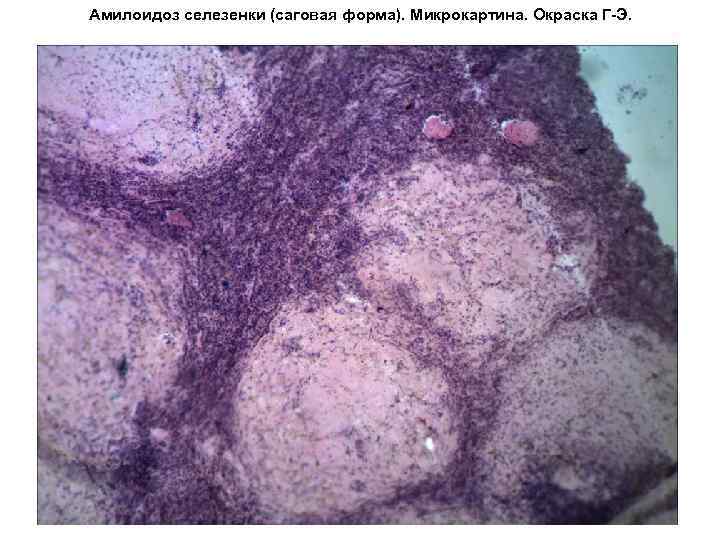
 Amiloidosi della milza (forma sago). Microimmagine. Colorazione G-E. Amiloidosi della milza (forma sago). Microimmagine. Colorazione G-E. (x240). (secondo V. A. Salimov) 1. follicoli di polpa bianca; 2. polpa rossa atrofizzata; 3. nuclei delle cellule sopravvissute; 4. depositi di amiloide nei follicoli della polpa bianca
Amiloidosi della milza (forma sago). Microimmagine. Colorazione G-E. Amiloidosi della milza (forma sago). Microimmagine. Colorazione G-E. (x240). (secondo V. A. Salimov) 1. follicoli di polpa bianca; 2. polpa rossa atrofizzata; 3. nuclei delle cellule sopravvissute; 4. depositi di amiloide nei follicoli della polpa bianca



 Amiloidosi della milza (forma grassa). Microimmagine. Colorazione G-E. Amiloidosi della milza (forma grassa). Microimmagine. Colorazione G-E. (x480). (secondo V. A. Salimov) 1. deposizione diffusa di amiloide; 2. polpa atrofizzata.
Amiloidosi della milza (forma grassa). Microimmagine. Colorazione G-E. Amiloidosi della milza (forma grassa). Microimmagine. Colorazione G-E. (x480). (secondo V. A. Salimov) 1. deposizione diffusa di amiloide; 2. polpa atrofizzata.
 Amiloidosi della milza (forma grassa). Microimmagine. Colorazione G-E.
Amiloidosi della milza (forma grassa). Microimmagine. Colorazione G-E.