Osteodistrofia ereditaria di Albright. Pseudoipoparatiroidismo (osteodistrofia ereditaria di Albright): difficoltà di ricerca diagnostica differenziale. Osservazione clinica Sintomi di pseudoipoparatiroidismo
Pseudoipoparatiroidismo
Cos'è lo pseudoipoparatiroidismo -
Pseudoipoparatiroidismo(Pseudē greco s falso + ipoparatiroidismo; sinonimo: osteodistrofia ereditaria di Albright, malattia di Albright) è una rara malattia ereditaria del sistema scheletrico, che simula l'ipoparatiroidismo e caratterizzata da un alterato metabolismo del calcio e del fosforo; spesso accompagnato da un ritardo nello sviluppo mentale e fisico.
Cosa provoca/cause dello pseudoipoparatiroidismo:
La causa dello pseudoipoparatiroidismo è un difetto congenito: insensibilità dei tessuti periferici all'azione del PTH.
Patogenesi (cosa succede?) durante lo pseudoipoparatiroidismo:
Si ritiene che lo pseudoipoparatiroidismo si basi sulla resistenza geneticamente determinata dei reni e dello scheletro all'azione dell'ormone paratiroideo a causa di un difetto nel complesso specifico citorecettore - ormone paratiroideo - adenilato ciclasi, che interrompe la formazione nei reni del ciclico 3 ", 5"-AMP, che è un mediatore intracellulare dell'azione dell'ormone paratiroideo sui processi metabolici . Lo pseudoipoparatiroidismo è una malattia geneticamente eterogenea. In alcuni pazienti, il citorecettore stesso che lega l'ormone paratiroideo è difettoso (pseudoipoparatiroidismo di tipo Ia); altri hanno un difetto nella proteina legante i nucleotidi, localizzata nel doppio strato lipidico della membrana cellulare e che lega funzionalmente il recettore all'adenilato ciclasi (tipo Ib pseudoipoparatiroidismo). Alcuni pazienti presentano un deficit enzimatico dell'adenilato ciclasi stessa (pseudoipoparatiroidismo di tipo II). Il deficit di cAMP derivante da questi difetti porta all'interruzione della sintesi di proteine specifiche che determinano l'effetto biologico dell'ormone paratiroideo. In questo modo si perde la sensibilità degli organi bersaglio, in particolare dei reni, all'ormone paratiroideo. Di conseguenza, l'escrezione di fosforo nelle urine diminuisce, si verifica iperfosfatemia e secondariamente si sviluppa ipocalcemia. Poiché nello pseudoipoparatiroidismo le ghiandole paratiroidi sono intatte, in risposta all'ipocalcemia può svilupparsi una secondaria, che stimola la produzione dell'ormone paratiroideo. iperparatiroidismo. L'aumento della formazione dell'ormone paratiroideo non causa un aumento dell'escrezione di fosforo e cAMP nelle urine a causa della resistenza geneticamente determinata dei tubuli renali all'ormone paratiroideo, ma è accompagnata da cambiamenti nel tessuto osseo caratteristici dell'iperparatiroidismo, che indica la conservazione della normale sensibilità degli osteoclasti all’ormone paratiroideo. Con lo pseudoipoparatiroidismo, l'attività della fosfatasi alcalina nel siero del sangue è aumentata o rientra nei limiti normali (0,5-1,3 µmol fosforo inorganico per 1 ml siero sanguigno per 1 H incubazione a 37°; definizione secondo Bodansky). Tutte le varianti dello pseudoipoparatiroidismo sono una malattia ereditaria, la natura dell'ereditarietà è autosomica dominante. La bassa fertilità degli uomini affetti da pseudoipoparatiroidismo spiega la rarità della sua trasmissione da padre in figlio; le donne si ammalano 2 volte più spesso degli uomini.
Di solito, con lo pseudoipoparatiroidismo, viene rilevata l'iperplasia compensatoria delle ghiandole paratiroidi (la presenza di adenomi in esse non è tipica). Nel tessuto osseo si notano cambiamenti tipici dell'iperparatiroidismo: osteoporosi diffusa, comparsa di cisti (i cosiddetti tumori bruni, tumori a cellule giganti). Il calcio rilasciato dalle ossa si deposita sotto forma di calcificazioni nel tessuto sottocutaneo, così come nei reni, nei muscoli, nel miocardio, nelle pareti delle grandi arterie, nella congiuntiva dell'occhio e lungo la periferia della cornea.
Sintomi dello pseudoipoparatiroidismo:
I segni clinici dello pseudoipoparatiroidismo sono simili a quelli dell'idiopatico ipoparatiroidismo. Ci sono attacchi di convulsioni toniche che si verificano spontaneamente o sotto l'influenza di sostanze irritanti. Le calcificazioni nel tessuto sottocutaneo tendono ad ulcerarsi. L'ossificazione sottocutanea è spesso grave al punto da imitare la miosite ossificante. I tratti caratteristici includono ritardo mentale, crescita stentata, faccia lunare, obesità e brachidattilia, in particolare accorciamento del primo, quarto e quinto metacarpo e metatarso. Si possono osservare esostosi multiple, discondroplasia e manifestazioni di iperparatiroidismo secondario sotto forma di riassorbimento sottoperiostale delle ossa delle dita; i cambiamenti nelle epifisi delle ossa sono gli stessi dell'osteodisplasia fibrosa. Si nota spesso vomito, così come ematuria dovuta alla formazione di calcoli di ossalato nel tratto urinario, vengono rilevate cataratta lenticolare e ipoplasia dello smalto dei denti.
Nei pazienti con pseudoipoparatiroidismo, insieme ad una diminuzione della sensibilità degli organi bersaglio all'ormone paratiroideo, si può osservare resistenza ad altri ormoni dipendenti dal sistema adenilato ciclasi, ad esempio le gonadi agli ormoni gonadotropici, la ghiandola tiroidea all'ormone stimolante la tiroide , organi bersaglio del glucagone e dell'ormone antidiuretico. C'è una maggiore frequenza Malattie autoimmuni E diabete mellito, si osservano ipotiroidismo e ipertiroidismo.
Esiste anche lo pseudopseudoipoparatiroidismo, caratterizzato dall'assenza di ipocalcemia, iperfosfatemia, convulsioni e osteomalacia.
Diagnosi di pseudoipoparatiroidismo:
La diagnosi nei casi tipici della malattia viene posta nei bambini di età compresa tra 5 e 10 anni sulla base di un quadro clinico caratteristico, anomalie multiple nello sviluppo dello scheletro osseo, presenza di ipocalcemia, iperfosfatemia, attività normale o aumentata della fosfatasi alcalina nel siero del sangue, ridotta escrezione di calcio e fosforo nelle urine, aumento dei livelli di ormone paratiroideo nel sangue. La presenza di resistenza tubulare renale all'ormone paratiroideo è confermata da un test basato sulla determinazione della quantità di fosfato e cAMP escreti nelle urine. L'assenza di un aumento significativo del contenuto di fosfati e cAMP nelle urine dopo la somministrazione dell'ormone paratiroideo al paziente indica una resistenza renale all'azione dell'ormone paratiroideo. Nei pazienti con ipoparatiroidismo idiopatico e postoperatorio, al contrario, dopo somministrazione endovenosa di 200 unità di ormone paratiroideo nelle urine, il contenuto di fosfati e cAMP entro 4 H aumenta 2-10 volte rispetto al livello iniziale. L'escrezione urinaria di idrossiprolina nei pazienti non trattati con pseudoipoparatiroidismo è normale o leggermente aumentata, mentre nell'ipoparatiroidismo è ridotta. La diagnosi a raggi X dello pseudoipoparatiroidismo si basa sull'identificazione di cambiamenti specifici nelle ossa e nei tessuti molli.
Lo pseudoipoparatiroidismo in combinazione con ipogonadismo nelle donne deve essere differenziato Shereshevskij - Sindrome di Turner, al quale lo pseudopseudoipoparatiroidismo è fenotipicamente simile. Nella sindrome di Shereshevskij-Turner la cromatina sessuale è assente; al posto delle ovaie si trovano filamenti di tessuto connettivo che non sono rilevabili durante l'esame rettale e l'ecografia.
Trattamento dello pseudoipoparatiroidismo:
Il trattamento dell'ipocalcemia consiste nel prescrivere integratori di calcio in dosi sufficienti a mantenere normali concentrazioni di calcio nel sangue. Di grande importanza è la terapia con vitamina D. La dose iniziale è calcolata a partire da 2000 UI/ kg peso corporeo al giorno, ma non più di 100.000 UI al giorno. Per evitare un sovradosaggio di preparati a base di vitamina D, è necessario monitorare la concentrazione di calcio nel sangue ogni 3-7 giorni durante le prime due settimane di trattamento e ogni mese per i successivi 2-3 mesi. Una volta raggiunta una concentrazione stabile di calcio nel sangue, è sufficiente controllarla una volta ogni 2-3 mesi. È possibile utilizzare calcitrina, diidrotachisterolo, oxidvit e altri preparati di forme attive di vitamina D. Una dieta con restrizione di fosforo aiuta a normalizzare la concentrazione di calcio nel sangue ed eliminare i sintomi dell'iperparatiroidismo secondario. In caso di insufficienza di altre ghiandole endocrine si effettua la terapia sostitutiva con ormoni appropriati. Il trattamento con l’ormone paratiroideo non è efficace. Per fermare gli attacchi convulsivi, viene somministrata per via endovenosa una soluzione al 10% di cloruro di calcio o gluconato di calcio; per via orale - soluzione al 5-10% di cloruro di calcio, 1 cucchiaio 3-4 volte al giorno: gluconato di calcio, lattato di calcio - fino a 10 G in un giorno.
La prognosi con la terapia razionale è favorevole. Considerando la natura ereditaria dello pseudoipoparatiroidismo, è necessaria la consulenza genetica medica riguardo alla possibilità di pseudoipoparatiroidismo nella prole.
Prevenzione dello pseudoipoparatiroidismo:
Quali medici dovresti contattare se soffri di pseudoipoparatiroidismo:
C'è qualcosa che ti preoccupa? Vuoi conoscere informazioni più dettagliate sullo pseudoipoparatiroidismo, le sue cause, i sintomi, i metodi di trattamento e prevenzione, il decorso della malattia e la dieta successiva? Oppure hai bisogno di un sopralluogo? Puoi fissare un appuntamento con un medico– clinica Eurolaboratorio sempre al tuo servizio! I migliori medici ti esamineranno, studieranno i segni esterni e ti aiuteranno a identificare la malattia dai sintomi, ti consiglieranno e forniranno l'assistenza necessaria e faranno una diagnosi. puoi anche tu chiamare un medico a casa. Clinica Eurolaboratorio aperto per te 24 ore su 24.
Come contattare la clinica:
Numero di telefono della nostra clinica a Kiev: (+38 044) 206-20-00 (multicanale). La segretaria della clinica selezionerà il giorno e l'ora convenienti per la visita dal medico. Sono indicate le nostre coordinate e indicazioni. Guarda più in dettaglio tutti i servizi della clinica su di esso.
(+38 044) 206-20-00
Se hai già effettuato delle ricerche in precedenza, Assicurati di portare i risultati a un medico per un consulto. Se gli studi non sono stati eseguiti, faremo tutto il necessario nella nostra clinica o con i nostri colleghi di altre cliniche.
Voi? È necessario adottare un approccio molto attento alla propria salute generale. Le persone non prestano abbastanza attenzione sintomi di malattie e non si rendono conto che queste malattie possono essere pericolose per la vita. Ci sono molte malattie che all'inizio non si manifestano nel nostro corpo, ma alla fine si scopre che, sfortunatamente, è troppo tardi per curarle. Ogni malattia ha i suoi segni specifici, manifestazioni esterne caratteristiche - le cosiddette sintomi della malattia. L’identificazione dei sintomi è il primo passo nella diagnosi delle malattie in generale. Per fare questo, devi solo farlo più volte all'anno. essere esaminato da un medico, al fine non solo di prevenire una terribile malattia, ma anche di mantenere uno spirito sano nel corpo e nell'organismo nel suo insieme.
Se vuoi fare una domanda a un medico, usa la sezione di consultazione online, forse lì troverai le risposte alle tue domande e leggi consigli per la cura di sé. Se sei interessato alle recensioni su cliniche e medici, prova a trovare le informazioni di cui hai bisogno nella sezione. Registrati anche sul portale medico Eurolaboratorio per tenerti aggiornato sulle ultime novità e aggiornamenti informativi sul sito, che ti verranno inviati automaticamente via email.
Altre malattie del gruppo Malattie del sistema endocrino, disturbi nutrizionali e disturbi metabolici:
| Crisi addisoniana (insufficienza surrenalica acuta) |
| Adenoma al seno |
| Distrofia adiposogenitale (malattia di Perchkranz-Babinski-Fröhlich) |
| Sindrome adrenogenitale |
| Acromegalia |
| Follia nutrizionale (distrofia nutrizionale) |
| Alcalosi |
| Alcaptonuria |
| Amiloidosi (distrofia amiloide) |
| Amiloidosi dello stomaco |
| Amiloidosi intestinale |
| Amiloidosi delle isole pancreatiche |
| Amiloidosi epatica |
| Amiloidosi dell'esofago |
| Acidosi |
| Malnutrizione proteico-energetica |
| Malattia delle cellule I (mucolipidosi di tipo II) |
| Malattia di Wilson-Konovalov (distrofia epatocerebrale) |
| Malattia di Gaucher (lipidosi da glucocerebroside, glucocerebrosidosi) |
| La malattia di Itsenko-Cushing |
| Malattia di Krabbe (leucodistrofia a cellule globoidi) |
| Malattia di Niemann-Pick (sfingomielinosi) |
| Malattia di Fabry |
| Gangliosidosi GM1 tipo I |
| Gangliosidosi GM1 tipo II |
| Gangliosidosi GM1 tipo III |
| Gangliosidosi GM2 |
| Gangliosidosi GM2 tipo I (idiozia amaurotica di Tay-Sachs, malattia di Tay-Sachs) |
| Gangliosidosi GM2 di tipo II (malattia di Sandhoff, idiozia amaurotica di Sandhoff) |
| Gangliosidosi GM2 giovanile |
| Gigantismo |
| Iperaldosteronismo |
| Iperaldosteronismo secondario |
| Iperaldosteronismo primario (sindrome di Conn) |
| Ipervitaminosi D |
| Ipervitaminosi A |
| Ipervitaminosi E |
| Ipervolemia |
| Coma iperglicemico (diabetico). |
| Iperkaliemia |
| Ipercalcemia |
| Iperlipoproteinemia di tipo I |
| Iperlipoproteinemia di tipo II |
| Iperlipoproteinemia di tipo III |
| Iperlipoproteinemia tipo IV |
| Iperlipoproteinemia tipo V |
| Coma iperosmolare |
| Iperparatiroidismo secondario |
| Iperparatiroidismo primario |
| Iperplasia del timo (ghiandola del timo) |
| Iperprolattinemia |
| Iperfunzione testicolare |
| Ipercolesterolemia |
| Ipovolemia |
| Coma ipoglicemico |
| Ipogonadismo |
| Ipogonadismo iperprolattinemico |
| Ipogonadismo isolato (idiopatico) |
| Ipogonadismo congenito primario (anorchismo) |
| Ipogonadismo acquisito primario |
| Ipokaliemia |
| Ipoparatiroidismo |
| Ipopituitarismo |
| Ipotiroidismo |
| Glicogenosi di tipo 0 (aglicogenosi) |
| Glicogenosi di tipo I (malattia di Gierke) |
| Glicogenosi di tipo II (malattia di Pompe) |
| Glicogenosi di tipo III (morbillo, malattia di Forbes, destrinosi limite) |
| Glicogenosi di tipo IV (morbo di Andersen, amilopectinosi, glicogenosi diffusa con cirrosi epatica) |
| Glicogenosi tipo IX (malattia di Haga) |
| Glicogenosi di tipo V (malattia di McArdle, deficit di miofosforilasi) |
| Glicogenosi di tipo VI (malattia di Hers, deficit di epatofosforilasi) |
| Glicogenosi di tipo VII (malattia di Tarui, deficit di miofosfofruttochinasi) |
| Glicogenosi di tipo VIII (morbo di Thomson) |
| Glicogenosi tipo XI |
| Glicogenosi tipo X |
| Carenza (insufficienza) di vanadio |
| Carenza di magnesio (insufficienza) |
| Carenza di manganese (insufficienza) |
| Carenza di rame (insufficienza) |
| Carenza (insufficienza) di molibdeno |
| Carenza (insufficienza) di cromo |
| Carenza di ferro |
| Carenza di calcio (carenza nutrizionale di calcio) |
| Carenza di zinco (carenza di zinco nella dieta) |
| Coma diabetico chetoacidotico |
| Disfunzione ovarica |
| Gozzo diffuso (endemico). |
| Pubertà ritardata |
| Eccesso di estrogeni |
| Involuzione delle ghiandole mammarie |
| Nanismo (bassa statura) |
| Kwashiorkor |
| Mastopatia cistica |
| Xantinuria |
| Coma acido lattico |
| Leucinosi (malattia dello sciroppo d’acero) |
| Lipidosi |
| Lipogranulomatosi di Farber |
| Lipodistrofia (degenerazione grassa) |
| Lipodistrofia generalizzata congenita (sindrome di Seyp-Lawrence) |
| Lipodistrofia ipermuscolare |
| Lipodistrofia post-iniezione |
| Lipodistrofia segmentaria progressiva |
| Lipomatosi |
| La lipomatosi è dolorosa |
| Leucodistrofia metacromatica |
| Coma mixedema |
E.V. Tozliyan, endocrinologo pediatrico, genetista, Ph.D., I.V. Shulyakova, neurologa, Ph.D.,
unità strutturale separata “Istituto clinico di ricerca di pediatria” dell'Istituto statale di istruzione di bilancio di istruzione professionale superiore “Università medica nazionale di ricerca russa intitolata a N.I. Pirogov" del Ministero della Sanità della Federazione Russa, Mosca
Parole chiave:
bambini, pseudoipoparatiroidismo, osteodistrofia ereditaria di Albright, obesità, ipocalcemia, diagnosi, resistenza all'ormone paratiroideo.
Parole chiave: bambini, pseudoipoparatiroidismo, osteodistrofia ereditaria di Albright, obesità, ipocalcemia, diagnostica, resistenza all'ormone paratiroideo.
Lo pseudoipoparatiroidismo (greco pseudes - falso + ipoparatiroidismo; sinonimo: osteodistrofia ereditaria di Albright, sindrome del pollo giavanese) è una rara malattia ereditaria del sistema scheletrico, che simula l'ipoparatiroidismo e caratterizzata da un alterato metabolismo del calcio e del fosforo; spesso accompagnato da un ritardo nello sviluppo mentale e fisico. La malattia fu descritta per la prima volta dall'endocrinologo americano Albright F. nel 1942. La prevalenza della malattia è di 7,9 per 1 milione di persone.
DATI GENETICI
Lo pseudoipoparatiroidismo (PHP) è una malattia geneticamente eterogenea. I dati sul tipo di trasmissione ereditaria sono contraddittori: sia i tipi dominanti legati all'X che quelli autosomici dominanti e autosomici recessivi. Nella maggior parte dei casi, lo sviluppo dell'osteodistrofia ereditaria di Albright è associato a mutazioni nel locus 20q13 del gene GNAS1 situato sul cromosoma 20 (Patten et al., 1990), che codifica per la proteina Gs-alfa associata al recettore dell'ormone paratiroideo (PTH). . Un fenotipo simile è stato rilevato anche in pazienti con delezione interstiziale del braccio lungo del cromosoma 2 nel locus 2q37.
PATOGENESI
La patogenesi dello pseudoipoparatiroidismo si basa sulla resistenza geneticamente determinata dei reni e dello scheletro all'azione dell'ormone paratiroideo a causa di un difetto nel complesso “citorecettore specifico – ormone paratiroideo – adenilato ciclasi”, che interrompe la formazione di 3”- ciclici , 5"-adenosina monofosfato (cAMP) nei reni, che è il mediatore intracellulare dell'azione dell'ormone paratiroideo sui processi metabolici. Lo pseudoipoparatiroidismo è geneticamente malattia eterogenea. In alcuni pazienti, il citorecettore stesso che lega l'ormone paratiroideo è difettoso (pseudoipoparatiroidismo di tipo 1A); altri hanno un difetto nella proteina legante i nucleotidi, localizzata nel doppio strato lipidico della membrana cellulare e che lega funzionalmente il recettore all'adenilato ciclasi (tipo 1B pseudoipoparatiroidismo). Alcuni pazienti presentano un deficit enzimatico dell'adenilato ciclasi stessa (pseudoipoparatiroidismo di tipo 2). Il deficit di cAMP derivante da questi difetti porta all'interruzione della sintesi di proteine specifiche che determinano l'effetto biologico dell'ormone paratiroideo. Pertanto, la sensibilità degli organi bersaglio all'ormone paratiroideo viene persa.
CARATTERISTICHE CLINICHE
Attualmente esistono 4 forme cliniche di patologia: tipi 1A, 1B, 1C e 2. La conoscenza delle loro caratteristiche cliniche e biochimiche e dei dati della ricerca genetica consente la diagnosi differenziale all'interno della forma nosologica stessa.
Segni comuni che fanno sospettare la malattia sono la sproporzionalità dello sviluppo fisico, la bassa statura (fino al nanismo) per accorciamento degli arti inferiori (foto 1), la brachidattilia (foto 2), la forma rotonda “a forma di luna” viso (foto 3). Talvolta si osservano esostosi e aplasia dentale.
Foto 1.
Aspetto di un bambino affetto da osteodistrofia di Albright
(caratteristiche del fenotipo, bassa statura per accorciamento degli arti inferiori)

Foto 2.
Caratteristiche del sistema scheletrico del paziente
con osteodistrofia di Albright
(brachidattilia - accorciamento delle dita)
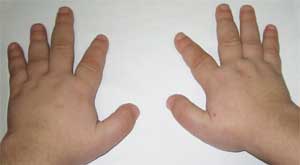
Foto 3.
Caratteristiche del fenotipo del bambino
con osteodistrofia di Albright
(viso tondo a forma di luna)
Un segno patognomonico è un forte accorciamento delle ossa metacarpali e metatarsali I, III e V (soprattutto III e IV), a seguito del quale le II dita delle mani e dei piedi sono più lunghe delle altre e quando la mano è serrata in un pugno, non ci sono rigonfiamenti nell'area delle articolazioni metacarpofalangee IV e V - il cosiddetto brachimetafalangismo. Vengono rilevate anche falangi larghe e corte, ispessimento della volta cranica e demineralizzazione delle ossa (osteoporosi) e obesità.
Il ritardo mentale (di solito di gravità moderata) si riscontra in circa il 20% dei pazienti. Secondo alcuni autori il ritardo mentale si manifesta nel 70% dei casi in presenza di ipocalcemia e nel 30% dei casi in normocalcemia. I processi mentali nei pazienti sono rallentati. Nello stato neurologico si notano spesso disagio motorio e reazioni nevrotiche: paure, ansia, irrequietezza, scarso sonno, aumento dei riflessi, convulsioni di natura tetanica causate dall'ipocalcemia e talvolta parossismi convulsivi. Sono stati descritti anche sintomi miopatici: affaticamento muscolare, debolezza muscolare. Si osservano spesso disturbi extrapiramidali: ipercinesia coreiforme, atetosi, emispasmo facciale, parkinsonismo; in alcuni casi si verificano parossismi epilettici, sintomi cerebellari: atassia, perdita di coordinazione.
Si riscontrano spesso calcificazioni dei tessuti molli e calcificazioni sottocutanee (torace, addome, tendini del tallone) e l'esame istologico delle stesse mostra osteoma cutaneo(Izraeli et al., 1992), cervello (gangli della base). È importante notare che le calcificazioni potrebbero già essere presenti alla nascita. Come risultato dell'ipocalcemia, di solito si sviluppa la cataratta e si verificano difetti dello smalto dei denti.
PSEUDOIPOPARATROOSI TIPO 1A
ha una modalità di trasmissione autosomica dominante. Il gene dello pseudoipoparatiroidismo di tipo 1A - GNAS1 - è localizzato sul braccio lungo del cromosoma 20, nel locus 20q13.2. Lo sviluppo della malattia è associato a una carenza della proteina legante i nucleotidi della guanina (proteina Gs). Allo stesso tempo, il PTH, legandosi ai recettori dei tessuti bersaglio, non è in grado di attivare l'adenosina monofosfato ciclico (cAMP) e provocare una risposta tissutale. Probabilmente, un meccanismo simile è alla base dello sviluppo di insensibilità dei tessuti di altri organi e ghiandole endocrine (ipofunzione della tiroide, delle gonadi, dell'ipofisi, del diabete mellito, nonché una ridotta risposta del fegato alla somministrazione di glucagone), osservato nello pseudoipoparatiroidismo tipo 1A. In questo tipo di patologia non si osserva il normale aumento dell'escrezione di cAMP nelle urine in risposta alla somministrazione esogena di PTH. La malattia viene diagnosticata più spesso all'età di 5-10 anni. I pazienti hanno bassa statura, collo corto, viso rotondo, accorciamento delle ossa metacarpali e metatarsali (di solito accorciamento del quarto e meno spesso del secondo dito) - il cosiddetto brachimetafalangismo. Si notano calcificazioni dei tessuti molli e calcificazioni sottocutanee, riscontrabili alla nascita; Si osserva spesso il coinvolgimento simultaneo di altre ghiandole endocrine: la tiroide (ipofunzione), le gonadi, il pancreas (diabete mellito). A causa dell'ipocalcemia si sviluppano spesso cataratta e difetti dello smalto dei denti. Come test diagnostico differenziale per distinguere il PHP di tipo 1A dall'ipoparatiroidismo: assenza di un effetto clinico dalla somministrazione parenterale di PTH sotto forma di aumento del livello di calcio nel sangue e aumento dell'escrezione renale di fosforo nelle urine (effetto fosforico).
L'esame biochimico rivela ipocalcemia, iperfosfatemia, aumento dei livelli di ormone paratiroideo nel sangue e ipofosfaturia. Il livello della proteina Gs nel sangue è ridotto. L'esame radiografico del sistema scheletrico rivela un accorciamento delle ossa metacarpali e metatarsali, una demineralizzazione generalizzata e un ispessimento delle ossa del calvario.
PSEUDOIPOPARATROOSI TIPO 1B
ha una trasmissione di tipo autosomico dominante, ma non è esclusa una trasmissione di tipo dominante legata all'X. È necessario tenere presente la penetranza incompleta talvolta osservata del gene della malattia e la possibilità di un portatore latente della patologia. Pertanto si raccomanda l'esame clinico (rilevamento del decorso subclinico della malattia) e biochimico (determinazione dei livelli di calcio, fosforo, PTH nel sangue) dei sospetti portatori della malattia. Il PHP di tipo 1B è causato da una carenza di recettori tissutali per l'ormone paratiroideo negli organi bersaglio e da una resistenza limitata all'ormone paratiroideo. Il quadro clinico è simile a quello del tipo 1A, ma non vi sono danni ad altre ghiandole endocrine e l'osteodistrofia è meno comune.
I pazienti non hanno una risposta renale alla somministrazione esogena dell'ormone paratiroideo sotto forma di un aumento dell'escrezione di adenosina monofosfato ciclico nelle urine; tuttavia, a differenza del tipo 1A, il livello della proteina Gs nel sangue è normale. Le donne sono colpite più spesso degli uomini, ma la gravità della malattia può essere la stessa sia negli uomini che nelle donne.
PSEUDOIPOPARATROOSI TIPO 1C
alcuni autori lo identificano con lo pseudo-pseudoipoparatiroidismo (PPHP), descritto da Albright F. nel 1952. È caratterizzato da un quadro clinico caratteristico del PHP, ma i livelli di calcio e fosforo nel sangue e nelle urine rimangono entro limiti normali. Anche i livelli di PTH e proteine Gs nel sangue rimangono a livelli normali. Alcuni pazienti con PHP di tipo 1C presentano eliminazioni de novo sul cromosoma 2. È possibile che questa variante della malattia sia un sottotipo del PHP di tipo 1A.
PSEUDOIPOPARATROOSI TIPO 2
clinicamente simile ad altri tipi di malattia, ma ha una modalità di trasmissione autosomica recessiva. Non si può escludere l'esistenza di forme patologiche autosomiche dominanti. La patogenesi dello sviluppo è associata alla resistenza intracellulare al cAMP. Il PTH si lega quindi ai recettori e provoca una normale risposta cellulare al PTH sotto forma di aumento dell'escrezione di cAMP. L’insensibilità intracellulare al cAMP, tuttavia, non consente di realizzare il pieno effetto del PTH. Allo stesso tempo, la normale reazione dei reni alla somministrazione esogena dell'ormone paratiroideo viene mantenuta sotto forma di maggiore escrezione di adenosina monofosfato ciclico nelle urine. È stato suggerito che il PHP di tipo 2 possa essere associato a carenza di vitamina D.
Pertanto, i tipi identificati di PGP sono clinicamente caratterizzati da una ridotta sensibilità degli organi bersaglio al PTH, ma differiscono nei meccanismi patogenetici della formazione dell'insensibilità tissutale.
DIAGNOSTICA
Un test diagnostico differenziale di laboratorio può essere il pattern dell'escrezione renale di cAMP in risposta alla somministrazione di PTH: si osserva un aumento dell'escrezione di cAMP nel tipo 2 e la sua assenza nel tipo 1. La diagnosi è confermata dal rilevamento di un livello ridotto di nucleotide guanina proteina legante (proteina Gs) nel sangue (in media 1,5–2 volte) rispetto alla norma. L'ipocalcemia è solitamente associata a iperfosfatemia e ipofosfaturia. I livelli di PTH sono elevati; nel tipo 1C, il livello di PTH è normale, il che dà origine al nome “pseudoipoparatiroidismo”. L'esame radiografico del sistema scheletrico rivela un accorciamento delle ossa metacarpali e metatarsali, spesso una demineralizzazione generalizzata (osteoporosi) e un ispessimento delle ossa della volta cranica. Il pattern dermatoglifico mostra uno spostamento assiale del triraggio palmare.
Criteri di diagnosi:
- bassa statura;
- viso tondo;
- ritardo nello sviluppo neuropsichico;
- anomalie scheletriche;
- basso calcio sierico;
- alto livello di ormone paratiroideo nel sangue;
- diminuzione dell’escrezione urinaria di fosfati e cAMP.
TRATTAMENTO E PREVENZIONE
Il trattamento dell'ipocalcemia consiste nel prescrivere integratori di calcio in dosi sufficienti a mantenere normali concentrazioni di calcio nel sangue. Di grande importanza è la terapia con vitamina D. Attualmente si utilizzano i metaboliti attivi della vitamina D: oxidvit, 1-alfa-D3, calcitrina, ecc. alla dose di 1-2 mcg/die con risultati positivi (aumento dei livelli di calcio nel sangue , diminuzione delle manifestazioni della sindrome convulsiva). Anche la tachistina (0,5-1,5 mg/die) è efficace. Questo farmaco aumenta l'assorbimento del calcio nell'intestino e quindi aiuta ad aumentare il livello di calcio nel sangue. La terapia anticonvulsivante viene utilizzata come trattamento aggiuntivo. Il trattamento non ha un effetto notevole sullo sviluppo intellettuale, ma insieme a una diminuzione dei sintomi della sindrome convulsiva, si osserva una regressione delle manifestazioni neurologiche (disturbi sottocorticali, ipercinesia coreiforme, atetosi, ecc.). Per evitare un sovradosaggio di preparati a base di vitamina D, è necessario monitorare la concentrazione di calcio nel sangue ogni 3-7 giorni durante le prime 2 settimane di trattamento e ogni mese per i successivi 2-3 mesi. Una volta raggiunta una concentrazione stabile di calcio nel sangue, è sufficiente controllarla una volta ogni 2-3 mesi. Una dieta povera di fosforo aiuta a normalizzare le concentrazioni di fosforo e calcio nel sangue ed eliminare i sintomi dell’iperparatiroidismo secondario. In caso di insufficienza di altre ghiandole endocrine si effettua la terapia sostitutiva con ormoni appropriati.
Il trattamento con l’ormone paratiroideo è inefficace. Per alleviare gli attacchi convulsivi, viene somministrata per via endovenosa una soluzione al 10% di cloruro di calcio o gluconato di calcio; per via orale – soluzione al 5–10% di cloruro di calcio, 1 cucchiaio 3–4 volte al giorno: gluconato di calcio, lattato di calcio – fino a 10 g al giorno.
PREVISIONE per la vita è determinata dalla gravità della sindrome convulsiva.
PREVENZIONE malattia si basa sui dati della consulenza genetica medica.
CONSULENZA MEDICO-GENETICA
Quando si effettua la consulenza genetica medica, si dovrebbe procedere dal tipo di ereditarietà autosomica dominante e dall'alto (50%) rischio di recidiva della malattia nella famiglia con forme ereditarie. Per identificare la natura del tipo di eredità, è necessario condurre un esame approfondito dei genitori, poiché la sindrome può manifestarsi con sintomi clinici minimi. Attualmente, la diagnosi genetica molecolare della malattia è stata sviluppata e viene migliorata mediante la tipizzazione delle mutazioni nel gene GNAS1 sul cromosoma 20. Sono in fase di sviluppo metodi per la diagnosi prenatale della malattia in generale e dei suoi tipi individuali.
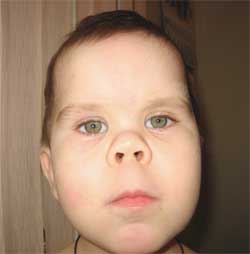
OSSERVAZIONE CLINICA Il ragazzo G., 14,5 anni (foto 4), è stato ricoverato presso l'Istituto Clinico di Ricerca di Pediatria con diagnosi di malattia degenerativa del sistema nervoso? idrocefalo esterno congenito; epilessia sintomatica; sindrome ereditaria? malattia da accumulo? encefalopatia metabolica; ipotiroidismo subclinico; bassa statura di origine mista; decadimento cognitivo.
Denunce, contestazioni al momento del ricovero intensi mal di testa parossistici localizzati nella regione frontale e accompagnati da vomito, che porta sollievo, diminuzione della memoria e del rendimento scolastico, attacchi convulsivi durante i quali si verificano contrazioni alla mano destra.
Foto 4.
Bambino G., 14,5 anni, affetto da osteodistrofia di Albright
(caratteristiche fenotipiche, bassa statura, arti accorciati, brachidattilia)

Storia famigliare: i genitori sono armeni di nazionalità, non imparentati con sangue e non presentano rischi professionali. Non sono stati rilevati casi di malattie mentali, epilessia o ritardi di sviluppo nel pedigree. Si dice che la sorella di Sibs, 17 anni, sia sana.
Storia della vita e della malattia: ragazzo della 2a gravidanza, avvenuta senza particolarità, secondo parto, a termine, fisiologico, peso alla nascita - 3100 g, lunghezza - 51 cm, ha gridato immediatamente, punteggio di Apgar - 7/9 punti. Peggioramento della condizione il 3° giorno – convulsioni neonatali, interrotte in maternità. Il primo periodo postnatale è privo di caratteristiche. Si è verificato un leggero ritardo nello sviluppo motorio nel primo anno di vita, camminando autonomamente da 1 anno e 3 mesi. In relazione a ciò, è stato osservato da un neurologo con una diagnosi di danno organico al sistema nervoso centrale; idrocefalo congenito; convulsioni neonatali; storia di convulsioni febbrili.
Ho ricevuto diacarb e finlepsina. Insorgenza degli attacchi a 1 anno e 11 mesi. – asimmetrico, tonico sotto forma di tensione al braccio e alla gamba destra, con occhi roteanti, fino a 2 minuti, senza perdita di coscienza, frequente fino a 10 episodi al giorno. Ho ricevuto Depakine in modo irregolare. Sullo sfondo del ritiro indipendente - un unico stato tonico. All'età di 2 anni, è stata eseguita una TC del cervello nel luogo di residenza, dove sono stati identificati singoli focolai di demielinizzazione nei lobi occipitali.
È stato consultato un neurochirurgo e gli è stato raccomandato un trattamento conservativo. Dall'età di 3 anni si osserva un ritardo nello sviluppo psico-linguistico e si consiglia l'osservazione da parte di uno psichiatra.
Dall'età di 4-5 anni, i genitori hanno iniziato a notare la deformazione e l'accorciamento delle dita delle mani e dei piedi, in particolare del 2°-4° dito simmetricamente su braccia e gambe, e una diminuzione degli indicatori di crescita. All'età di 8 anni, la conclusione del logopedista è un disturbo generale del linguaggio di 2°-3° livello; si raccomanda l'istruzione in una scuola specializzata. Alla stessa età, esame da parte di un genetista nel luogo di residenza, conclusione: malattia metabolica ereditaria? È stato raccomandato uno studio sugli aminoacidi nel sangue; non sono stati rilevati cambiamenti; conclusione finale: non è stata identificata alcuna evidenza di una malattia metabolica ereditaria; ipocondroplasia; Si raccomanda il trattamento da parte di un neurologo ed endocrinologo.
Tavolo.
Profilo dello sviluppo mentale del bambino G., 14,5 anni (QI = 68)

All'età di 8 anni, è stato consultato da un endocrinologo riguardo al ritardo della crescita e dello sviluppo. Un esame radiografico delle mani ha rivelato le seguenti caratteristiche: le falangi medie e principali e le ossa metacarpali erano accorciate e ispessite; La diagnosi del radiologo era acondroplasia.
Esaminato ripetutamente nel luogo di residenza in un ospedale neurologico. All'età di 12 anni, sono comparsi attacchi convulsivi senza perdita di coscienza con contrazioni del braccio destro, di natura seriale, è stata prescritta una terapia anticonvulsivante (Depakine) e la frequenza degli attacchi è diminuita significativamente. All'età di 13 anni, è stata eseguita una risonanza magnetica del cervello con contrasto: cambiamenti simmetrici nella base dei lobi temporali a livello dei nuclei sotto forma di un aumento del segnale MR, tipico del tossico (manganese) o encefalopatie metaboliche (rame, ferro).
Riesaminato all'età di 13 anni 3 mesi. da un endocrinologo, uno studio del profilo tiroideo ha rivelato un aumento dell'ormone stimolante la tiroide (TSH), è stato diagnosticato un ipotiroidismo subclinico e gli è stata prescritta L-tiroxina.
Analizzando la scheda ambulatoriale del bambino e la documentazione nel luogo di residenza, è stato effettuato uno studio del calcio e del fosforo una volta, all'età di 1,5 anni, è stata notata ipocalcemia, ma in questa occasione non è stato effettuato alcun ulteriore esame. Considerando l'incertezza della diagnosi nel luogo di residenza, il genetista ha inviato il bambino a Mosca, all'Istituto clinico di ricerca scientifica di pediatria, per chiarire la diagnosi.
Dati oggettivi della ricerca:
Altezza – 143 cm, peso – 43 kg.
Lo sviluppo fisico è molto basso, armonico, il fisico è sproporzionato per l'accorciamento degli arti. La Sds di crescita corrisponde a –2,8 deviazioni dalla norma (norma –2+2).
Caratteristiche fenotipiche: viso tondo, collo corto, rime palpebrali antimongoloidi, ponte nasale largo, fronte alta, brachidattilia, accorciamento delle ossa metacarpali IV e V e metatarsali (foto 5). Organi interni – senza alcuna particolarità. Sviluppo sessuale – Stadio di Tanner III–IV (che corrisponde all’età).
Dati di laboratorio e di ricerca funzionale:
L'analisi clinica del sangue e delle urine è normale.
Esame del sangue biochimico: calcio totale – 1,39 (normale 2,02–2,6 mmol/l), calcio ionizzato – 0,61 (normale 1,13–1,32 mmol/l), fosforo inorganico – 3,66 (normale 0,86–1,56 mmol/l), altri indicatori rientrano nei limiti limiti normali.
Analisi biochimica delle urine: l'escrezione renale dei fosfati è ridotta - 11,5 mmol/l (normale 19–32 mmol/l).
Profilo tiroideo: TSH – 11,75 (intervallo normale 0,4–4,0 µIU/ml), T4 libera – 0,49 (intervallo normale 1,0–1,8 ng/dL).
Ormone paratiroideo – 499 (normale 12–65 pg/ml), STH – 7 ng/ml (normale 7–10 ng/ml), somatomedina-C – 250 ng/ml (normale 88–360 ng/ml).
Ultrasuoni degli organi interni - senza alcuna caratteristica.
ECG – migrazione del pacemaker sopraventricolare sullo sfondo di una frequenza cardiaca regolare di 71–80 battiti/min. Blocco incompleto della branca destra. Interruzione del processo di ripolarizzazione nel miocardio della parete posteriore del ventricolo sinistro (diminuzione di ZT III, aVF).
R-grafia della colonna vertebrale - scoliosi destra della colonna vertebrale toracica di 1o grado, grave osteoporosi.
R-grafia delle mani con cattura degli avambracci - accorciamento e allargamento delle falangi terminali e medie. Età ossea: 13,5-14 anni.
Non sono stati registrati modelli EEG di attività epilettica.
MRI del cervello - Immagine RM di più focolai sottocorticali di aumento del segnale MR nei lobi frontali, idrocefalo compensato esterno con atrofia della sostanza cerebrale.
La MSCT del cervello mostra aree simmetriche di calcificazione dei nuclei lentiformi. Aree iperdense diffuse nel talamo, nuclei caudati con area di calcificazione a destra. Calcificazioni puntiformi multiple dei tessuti molli tegumentari del cranio.
Audiogramma – senza patologia.
La diagnostica del DNA nel gene GNAS1 è in corso.
Consulenze specialistiche:
Endocrinologo – Osteodistrofia ereditaria di Albright tipo 1A (pseudoipoparatiroidismo). Ipotiroidismo primario, compenso farmacologico incompleto.
Oculista – cataratta secondaria completa. Si consiglia il trattamento chirurgico.
Psicologo – deterioramento cognitivo (il profilo psicologico del bambino è presentato nella tabella).
Tenendo conto del fenotipo del bambino, dell'anamnesi medica, dei risultati di ulteriori studi (ipocalcemia, iperfosfatemia, ipofosfaturia, aumento dell'ormone paratiroideo nel sangue), calcificazioni nel cervello, presenza di cataratta, ipotiroidismo), è stata fatta una diagnosi: osteodistrofia ereditaria di Albright tipo 1A (pseudoipoparatiroidismo). Si consiglia di eseguire la diagnostica del DNA, cercando mutazioni nel gene GNAS1.
Trattamento: Si consiglia al bambino di assumere eutirox alla dose di 100 mcg/die; metabolita attivo della vitamina D - alfa-D3 (Teva) alla dose di 2 mcg/giorno; calcio (Sandoz) 2000 mg/giorno; uso costante della terapia anticonvulsivante - finlepsina 800 mg/giorno sotto la supervisione di un neurologo-epilettologo; lezioni con un logopedista e uno psicologo; terapia energetico-tropica (Elkar e coenzima Q10 in dosi correlate all'età). Indicatori di monitoraggio del metabolismo fosfo-calcio, livelli di ormone paratiroideo.
Così, L'osservazione clinica presentata dimostra la complessità della ricerca diagnostica differenziale, l'importanza dello studio tempestivo di semplici parametri biochimici (in caso di epilessia è richiesto uno screening ripetuto degli indicatori del metabolismo fosforo-calcio), i risultati della diagnosi tardiva di una malattia geneticamente determinata , la necessità di integrare i segni individuali nel fenotipo generale di una particolare condizione patologica per la diagnosi tempestiva e mirata di alcune forme di malattie ereditarie. La diagnosi tempestiva e il chiarimento della genesi di ciascuna sindrome sono particolarmente importanti, poiché consentono di trovare l'approccio ottimale al trattamento di queste condizioni e alla prevenzione di possibili complicanze (fino alla disabilità del bambino); prevenzione delle recidive di malattie ereditarie nelle famiglie colpite (consulenza medica e genetica). Ciò impone la necessità che i medici di varie specialità navighino chiaramente nel flusso della patologia determinata ereditariamente. L'elenco dei riferimenti è in redazione.
Pseudoipoparatiroidismo(Pseudē greco s falso + ipoparatiroidismo; sinonimo: osteodistrofia ereditaria di Albright, malattia di Albright) è una rara malattia ereditaria del sistema scheletrico, che simula l'ipoparatiroidismo e caratterizzata da un alterato metabolismo del calcio e del fosforo; spesso accompagnato da un ritardo nello sviluppo mentale e fisico.
Cosa provoca/cause dello pseudoipoparatiroidismo:
La causa dello pseudoipoparatiroidismo è un difetto congenito: insensibilità dei tessuti periferici all'azione del PTH.
Patogenesi (cosa succede?) durante lo pseudoipoparatiroidismo:
Si ritiene che lo pseudoipoparatiroidismo si basi sulla resistenza geneticamente determinata dei reni e dello scheletro all'azione dell'ormone paratiroideo a causa di un difetto nel complesso specifico citorecettore - ormone paratiroideo - adenilato ciclasi, che interrompe la formazione nei reni del ciclico 3 ", 5"-AMP, che è un mediatore intracellulare dell'azione dell'ormone paratiroideo sui processi metabolici . Lo pseudoipoparatiroidismo è una malattia geneticamente eterogenea. In alcuni pazienti, il citorecettore stesso che lega l'ormone paratiroideo è difettoso (pseudoipoparatiroidismo di tipo Ia); altri hanno un difetto nella proteina legante i nucleotidi, localizzata nel doppio strato lipidico della membrana cellulare e che lega funzionalmente il recettore all'adenilato ciclasi (tipo Ib pseudoipoparatiroidismo). Alcuni pazienti presentano un deficit enzimatico dell'adenilato ciclasi stessa (pseudoipoparatiroidismo di tipo II). Il deficit di cAMP derivante da questi difetti porta all'interruzione della sintesi di proteine specifiche che determinano l'effetto biologico dell'ormone paratiroideo. In questo modo si perde la sensibilità degli organi bersaglio, in particolare dei reni, all'ormone paratiroideo. Di conseguenza, l'escrezione di fosforo nelle urine diminuisce, si verifica iperfosfatemia e secondariamente si sviluppa ipocalcemia. Poiché nello pseudoipoparatiroidismo le ghiandole paratiroidi sono intatte, in risposta all'ipocalcemia può svilupparsi una secondaria, che stimola la produzione dell'ormone paratiroideo. iperparatiroidismo. L'aumento della formazione dell'ormone paratiroideo non causa un aumento dell'escrezione di fosforo e cAMP nelle urine a causa della resistenza geneticamente determinata dei tubuli renali all'ormone paratiroideo, ma è accompagnata da cambiamenti nel tessuto osseo caratteristici dell'iperparatiroidismo, che indica la conservazione della normale sensibilità degli osteoclasti all’ormone paratiroideo. Con lo pseudoipoparatiroidismo, l'attività della fosfatasi alcalina nel siero del sangue è aumentata o rientra nei limiti normali (0,5-1,3 µmol fosforo inorganico per 1 ml siero sanguigno per 1 H incubazione a 37°; definizione secondo Bodansky). Tutte le varianti dello pseudoipoparatiroidismo sono una malattia ereditaria, la natura dell'ereditarietà è autosomica dominante. La bassa fertilità degli uomini affetti da pseudoipoparatiroidismo spiega la rarità della sua trasmissione da padre in figlio; le donne si ammalano 2 volte più spesso degli uomini.
Di solito, con lo pseudoipoparatiroidismo, viene rilevata l'iperplasia compensatoria delle ghiandole paratiroidi (la presenza di adenomi in esse non è tipica). Nel tessuto osseo si notano cambiamenti tipici dell'iperparatiroidismo: osteoporosi diffusa, comparsa di cisti (i cosiddetti tumori bruni, tumori a cellule giganti). Il calcio rilasciato dalle ossa si deposita sotto forma di calcificazioni nel tessuto sottocutaneo, così come nei reni, nei muscoli, nel miocardio, nelle pareti delle grandi arterie, nella congiuntiva dell'occhio e lungo la periferia della cornea.
Sintomi dello pseudoipoparatiroidismo:
I segni clinici dello pseudoipoparatiroidismo sono simili a quelli dell'idiopatico ipoparatiroidismo. Ci sono attacchi di convulsioni toniche che si verificano spontaneamente o sotto l'influenza di sostanze irritanti. Le calcificazioni nel tessuto sottocutaneo tendono ad ulcerarsi. L'ossificazione sottocutanea è spesso grave al punto da imitare la miosite ossificante. I tratti caratteristici includono ritardo mentale, crescita stentata, faccia lunare, obesità e brachidattilia, in particolare accorciamento del primo, quarto e quinto metacarpo e metatarso. Si possono osservare esostosi multiple, discondroplasia e manifestazioni di iperparatiroidismo secondario sotto forma di riassorbimento sottoperiostale delle ossa delle dita; i cambiamenti nelle epifisi delle ossa sono gli stessi dell'osteodisplasia fibrosa. Si nota spesso vomito, così come ematuria dovuta alla formazione di calcoli di ossalato nel tratto urinario, vengono rilevate cataratta lenticolare e ipoplasia dello smalto dei denti.
Nei pazienti con pseudoipoparatiroidismo, insieme ad una diminuzione della sensibilità degli organi bersaglio all'ormone paratiroideo, si può osservare resistenza ad altri ormoni dipendenti dal sistema adenilato ciclasi, ad esempio le gonadi agli ormoni gonadotropici, la ghiandola tiroidea all'ormone stimolante la tiroide , organi bersaglio del glucagone e dell'ormone antidiuretico. C'è una maggiore frequenza Malattie autoimmuni E diabete mellito, si osservano ipotiroidismo e ipertiroidismo.
Esiste anche lo pseudopseudoipoparatiroidismo, caratterizzato dall'assenza di ipocalcemia, iperfosfatemia, convulsioni e osteomalacia.
Diagnosi di pseudoipoparatiroidismo:
La diagnosi nei casi tipici della malattia viene posta nei bambini di età compresa tra 5 e 10 anni sulla base di un quadro clinico caratteristico, anomalie multiple nello sviluppo dello scheletro osseo, presenza di ipocalcemia, iperfosfatemia, attività normale o aumentata della fosfatasi alcalina nel siero del sangue, ridotta escrezione di calcio e fosforo nelle urine, aumento dei livelli di ormone paratiroideo nel sangue. La presenza di resistenza tubulare renale all'ormone paratiroideo è confermata da un test basato sulla determinazione della quantità di fosfato e cAMP escreti nelle urine. L'assenza di un aumento significativo del contenuto di fosfati e cAMP nelle urine dopo la somministrazione dell'ormone paratiroideo al paziente indica una resistenza renale all'azione dell'ormone paratiroideo. Nei pazienti con ipoparatiroidismo idiopatico e postoperatorio, al contrario, dopo somministrazione endovenosa di 200 unità di ormone paratiroideo nelle urine, il contenuto di fosfati e cAMP entro 4 H aumenta 2-10 volte rispetto al livello iniziale. L'escrezione urinaria di idrossiprolina nei pazienti non trattati con pseudoipoparatiroidismo è normale o leggermente aumentata, mentre nell'ipoparatiroidismo è ridotta. La diagnosi a raggi X dello pseudoipoparatiroidismo si basa sull'identificazione di cambiamenti specifici nelle ossa e nei tessuti molli.
Lo pseudoipoparatiroidismo in combinazione con ipogonadismo nelle donne deve essere differenziato Shereshevskij - Sindrome di Turner, al quale lo pseudopseudoipoparatiroidismo è fenotipicamente simile. Nella sindrome di Shereshevskij-Turner la cromatina sessuale è assente; al posto delle ovaie si trovano filamenti di tessuto connettivo che non sono rilevabili durante l'esame rettale e l'ecografia.
Trattamento dello pseudoipoparatiroidismo:
Il trattamento dell'ipocalcemia consiste nel prescrivere integratori di calcio in dosi sufficienti a mantenere normali concentrazioni di calcio nel sangue. Di grande importanza è la terapia con vitamina D. La dose iniziale è calcolata a partire da 2000 UI/ kg peso corporeo al giorno, ma non più di 100.000 UI al giorno. Per evitare un sovradosaggio di preparati a base di vitamina D, è necessario monitorare la concentrazione di calcio nel sangue ogni 3-7 giorni durante le prime due settimane di trattamento e ogni mese per i successivi 2-3 mesi. Una volta raggiunta una concentrazione stabile di calcio nel sangue, è sufficiente controllarla una volta ogni 2-3 mesi. È possibile utilizzare calcitrina, diidrotachisterolo, oxidvit e altri preparati di forme attive di vitamina D. Una dieta con restrizione di fosforo aiuta a normalizzare la concentrazione di calcio nel sangue ed eliminare i sintomi dell'iperparatiroidismo secondario. In caso di insufficienza di altre ghiandole endocrine si effettua la terapia sostitutiva con ormoni appropriati. Il trattamento con l’ormone paratiroideo non è efficace. Per fermare gli attacchi convulsivi, viene somministrata per via endovenosa una soluzione al 10% di cloruro di calcio o gluconato di calcio; per via orale - soluzione al 5-10% di cloruro di calcio, 1 cucchiaio 3-4 volte al giorno: gluconato di calcio, lattato di calcio - fino a 10 G in un giorno.
La prognosi con la terapia razionale è favorevole. Considerando la natura ereditaria dello pseudoipoparatiroidismo, è necessaria la consulenza genetica medica riguardo alla possibilità di pseudoipoparatiroidismo nella prole.
Prevenzione dello pseudoipoparatiroidismo:
Quali medici dovresti contattare se soffri di pseudoipoparatiroidismo:
C'è qualcosa che ti preoccupa? Vuoi conoscere informazioni più dettagliate sullo pseudoipoparatiroidismo, le sue cause, i sintomi, i metodi di trattamento e prevenzione, il decorso della malattia e la dieta successiva? Oppure hai bisogno di un sopralluogo? Puoi fissare un appuntamento con un medico– clinica Eurolaboratorio sempre al tuo servizio! I migliori medici ti esamineranno, studieranno i segni esterni e ti aiuteranno a identificare la malattia dai sintomi, ti consiglieranno e forniranno l'assistenza necessaria e faranno una diagnosi. puoi anche tu chiamare un medico a casa. Clinica Eurolaboratorio aperto per te 24 ore su 24.
Come contattare la clinica:
Numero di telefono della nostra clinica a Kiev: (+38 044) 206-20-00 (multicanale). La segretaria della clinica selezionerà il giorno e l'ora convenienti per la visita dal medico. Sono indicate le nostre coordinate e indicazioni. Guarda più in dettaglio tutti i servizi della clinica su di esso.
(+38 044) 206-20-00
Se hai già effettuato delle ricerche in precedenza, Assicurati di portare i risultati a un medico per un consulto. Se gli studi non sono stati eseguiti, faremo tutto il necessario nella nostra clinica o con i nostri colleghi di altre cliniche.
Voi? È necessario adottare un approccio molto attento alla propria salute generale. Le persone non prestano abbastanza attenzione sintomi di malattie e non si rendono conto che queste malattie possono essere pericolose per la vita. Ci sono molte malattie che all'inizio non si manifestano nel nostro corpo, ma alla fine si scopre che, sfortunatamente, è troppo tardi per curarle. Ogni malattia ha i suoi segni specifici, manifestazioni esterne caratteristiche - le cosiddette sintomi della malattia. L’identificazione dei sintomi è il primo passo nella diagnosi delle malattie in generale. Per fare questo, devi solo farlo più volte all'anno. essere esaminato da un medico, al fine non solo di prevenire una terribile malattia, ma anche di mantenere uno spirito sano nel corpo e nell'organismo nel suo insieme.
Se vuoi fare una domanda a un medico, usa la sezione di consultazione online, forse lì troverai le risposte alle tue domande e leggi consigli per la cura di sé. Se sei interessato alle recensioni su cliniche e medici, prova a trovare le informazioni di cui hai bisogno nella sezione. Registrati anche sul portale medico Eurolaboratorio per tenerti aggiornato sulle ultime novità e aggiornamenti informativi sul sito, che ti verranno inviati automaticamente via email.
Altre malattie del gruppo Malattie del sistema endocrino, disturbi nutrizionali e disturbi metabolici:
| Crisi addisoniana (insufficienza surrenalica acuta) |
| Adenoma al seno |
| Distrofia adiposogenitale (malattia di Perchkranz-Babinski-Fröhlich) |
| Sindrome adrenogenitale |
| Acromegalia |
| Follia nutrizionale (distrofia nutrizionale) |
| Alcalosi |
| Alcaptonuria |
| Amiloidosi (distrofia amiloide) |
| Amiloidosi dello stomaco |
| Amiloidosi intestinale |
| Amiloidosi delle isole pancreatiche |
| Amiloidosi epatica |
| Amiloidosi dell'esofago |
| Acidosi |
| Malnutrizione proteico-energetica |
| Malattia delle cellule I (mucolipidosi di tipo II) |
| Malattia di Wilson-Konovalov (distrofia epatocerebrale) |
| Malattia di Gaucher (lipidosi da glucocerebroside, glucocerebrosidosi) |
| La malattia di Itsenko-Cushing |
| Malattia di Krabbe (leucodistrofia a cellule globoidi) |
| Malattia di Niemann-Pick (sfingomielinosi) |
| Malattia di Fabry |
| Gangliosidosi GM1 tipo I |
| Gangliosidosi GM1 tipo II |
| Gangliosidosi GM1 tipo III |
| Gangliosidosi GM2 |
| Gangliosidosi GM2 tipo I (idiozia amaurotica di Tay-Sachs, malattia di Tay-Sachs) |
| Gangliosidosi GM2 di tipo II (malattia di Sandhoff, idiozia amaurotica di Sandhoff) |
| Gangliosidosi GM2 giovanile |
| Gigantismo |
| Iperaldosteronismo |
| Iperaldosteronismo secondario |
| Iperaldosteronismo primario (sindrome di Conn) |
| Ipervitaminosi D |
| Ipervitaminosi A |
| Ipervitaminosi E |
| Ipervolemia |
| Coma iperglicemico (diabetico). |
| Iperkaliemia |
| Ipercalcemia |
| Iperlipoproteinemia di tipo I |
| Iperlipoproteinemia di tipo II |
| Iperlipoproteinemia di tipo III |
| Iperlipoproteinemia tipo IV |
| Iperlipoproteinemia tipo V |
| Coma iperosmolare |
| Iperparatiroidismo secondario |
| Iperparatiroidismo primario |
| Iperplasia del timo (ghiandola del timo) |
| Iperprolattinemia |
| Iperfunzione testicolare |
| Ipercolesterolemia |
| Ipovolemia |
| Coma ipoglicemico |
| Ipogonadismo |
| Ipogonadismo iperprolattinemico |
| Ipogonadismo isolato (idiopatico) |
| Ipogonadismo congenito primario (anorchismo) |
| Ipogonadismo acquisito primario |
| Ipokaliemia |
| Ipoparatiroidismo |
| Ipopituitarismo |
| Ipotiroidismo |
| Glicogenosi di tipo 0 (aglicogenosi) |
| Glicogenosi di tipo I (malattia di Gierke) |
| Glicogenosi di tipo II (malattia di Pompe) |
| Glicogenosi di tipo III (morbillo, malattia di Forbes, destrinosi limite) |
| Glicogenosi di tipo IV (morbo di Andersen, amilopectinosi, glicogenosi diffusa con cirrosi epatica) |
| Glicogenosi tipo IX (malattia di Haga) |
| Glicogenosi di tipo V (malattia di McArdle, deficit di miofosforilasi) |
| Glicogenosi di tipo VI (malattia di Hers, deficit di epatofosforilasi) |
| Glicogenosi di tipo VII (malattia di Tarui, deficit di miofosfofruttochinasi) |
| Glicogenosi di tipo VIII (morbo di Thomson) |
| Glicogenosi tipo XI |
| Glicogenosi tipo X |
| Carenza (insufficienza) di vanadio |
| Carenza di magnesio (insufficienza) |
| Carenza di manganese (insufficienza) |
| Carenza di rame (insufficienza) |
| Carenza (insufficienza) di molibdeno |
| Carenza (insufficienza) di cromo |
| Carenza di ferro |
| Carenza di calcio (carenza nutrizionale di calcio) |
| Carenza di zinco (carenza di zinco nella dieta) |
| Coma diabetico chetoacidotico |
| Disfunzione ovarica |
| Gozzo diffuso (endemico). |
| Pubertà ritardata |
| Eccesso di estrogeni |
| Involuzione delle ghiandole mammarie |
| Nanismo (bassa statura) |
| Kwashiorkor |
| Mastopatia cistica |
| Xantinuria |
| Coma acido lattico |
| Leucinosi (malattia dello sciroppo d’acero) |
| Lipidosi |
| Lipogranulomatosi di Farber |
| Lipodistrofia (degenerazione grassa) |
| Lipodistrofia generalizzata congenita (sindrome di Seyp-Lawrence) |
| Lipodistrofia ipermuscolare |
| Lipodistrofia post-iniezione |
| Lipodistrofia segmentaria progressiva |
| Lipomatosi |
| La lipomatosi è dolorosa |
| Leucodistrofia metacromatica |
| Coma mixedema |
| Fibrosi cistica (fibrosi cistica) |
Lo pseudoipoparatiroidismo è una malattia abbastanza rara del sistema scheletrico, la cui essenza è una violazione del metabolismo del calcio e del fosforo. Il paziente di solito sperimenta l'inibizione dello sviluppo mentale e fisico. La malattia è ereditaria.
Dal fatto che il nome della malattia ha il prefisso “pseudo”, si capisce facilmente che imita l'ipoparatiroidismo. Il secondo nome di questa malattia è osteodistrofia ereditaria di Albright, dal nome del medico che studiò e descrisse questa patologia a metà del secolo scorso.
Tipi di pseudoipoparatiroidismo
Esistono due tipi di pseudoipoparatiroidismo, a seconda che il livello di calcio nel sangue sia cambiato o meno. Il primo tipo di malattia di Albright presenta gli stessi segni clinici dell'ipoparatiroidismo idiopatico ed è caratterizzato da una diminuzione dei livelli di calcio nel sangue. Non esiste sensibilità tissutale all’ormone paratiroideo. Il secondo tipo di pseudoipoparatiroidismo, al contrario, differisce in quanto il livello di calcio è normale. Pertanto, questo tipo è chiamato pseudopseudoipoparatiroidismo. A proposito, secondo gli endocrinologi, una forma può facilmente trasformarsi in un'altra e i membri della stessa famiglia possono avere diversi tipi di malattia.
Cause dello pseudoipoparatiroidismo
I disturbi del metabolismo del fosforo-calcio si sviluppano a causa della resistenza dei tessuti all'ormone paratiroideo, prodotto dalle ghiandole paratiroidi. Sulla base dello pseudoipoparatiroidismo è stato confermato per la prima volta il fenomeno della ridotta sensibilità dei tessuti all'ormone prodotto dalle ghiandole endocrine o introdotto dall'esterno.
Lo pseudoipoparatiroidismo è una patologia genetica. È causato da una sindrome congenita - un recettore cellulare specifico - l'ormone paratiroideo-adenilato ciclasi, motivo per cui i tessuti periferici perdono la sensibilità all'ormone paratiroideo.
Chi è più suscettibile allo pseudoipoparatiroidismo? Innanzitutto i figli del paziente e gli altri parenti, poiché la malattia è ereditaria, autosomica dominante. Le donne soffrono più spesso della malattia di Albright. Gli esperti lo spiegano con il fatto che gli uomini con pseudoipoparatiroidismo hanno una bassa fertilità e l'ereditarietà stessa viene trasmessa di padre in figlio solo in casi isolati.
Non ci sono statistiche sulla diffusione di questa malattia, nei lavori medici sono descritti circa trecento casi.
Sintomi di pseudoipoparatiroidismo
- Anche i problemi agli organi del sistema endocrino con pseudoipoparatiroidismo causano i sintomi principali: il paziente è solitamente tozzo, più basso degli altri a causa degli arti inferiori accorciati, è obeso, il livello di glucosio nel sangue è elevato, il viso assume un aspetto lunare forma sagomata;
- Nel gentil sesso, il ciclo mestruale viene interrotto. Spesso questi segni sono accompagnati da acromegalia, ginecomastia, vari processi autoimmuni, sindrome di Itsenko-Cushing e altri disturbi. In questi pazienti anche il rischio di diabete mellito e diabete insipido è maggiore;
- Chi soffre della malattia di Albright lamenta spesso problemi ai denti (lo smalto è danneggiato); i pazienti sono anche disturbati da vomito, convulsioni toniche - sia spontanee che insorte sotto l'influenza di sostanze irritanti; si può osservare l'urina nel sangue. C'è il rischio di cataratta. Caratterizzato da ritardo mentale. I bambini con questa malattia non riescono a far fronte al programma scolastico, la loro memoria è ridotta;
- Cambiamenti significativi si verificano anche nel sistema osteoarticolare: appare l'osteoporosi diffusa, si sviluppano cisti (tumori marroni). I pazienti con pseudoipoparatiroidismo hanno maggiori probabilità di soffrire di fratture o deformità ossee. Il primo, quarto e quinto metacarpo e metatarso sono accorciati e si osservano segni di riassorbimento osseo sulle mani e sui piedi. Inoltre, il calcio, che viene rilasciato dalle ossa, comincia ad accumularsi nel tessuto sottocutaneo, formando calcificazioni (questo accade anche con la miosite ossificante), nella zona delle articolazioni e nella cavità cranica. Le calcificazioni sono presenti anche nei muscoli, nei reni, nel miocardio e sulle pareti delle grandi arterie;
- Un'altra caratteristica della malattia di Albright è la pigmentazione della pelle. Macchie marroni sparse su tutto il corpo (sia su un lato che sull'altro).
Per quanto riguarda la capacità lavorativa, dipende dalla gravità del processo e dall'efficacia del trattamento farmacologico. Se la forma della malattia è latente e non si verificano attacchi evidenti, la capacità lavorativa è parzialmente preservata. Ma ci sono una serie di restrizioni: in particolare, non è possibile lavorare con meccanismi di movimento o nei trasporti, sono vietati anche il sovraccarico neuropsichico e l'eccessivo lavoro fisico. Se ci sono frequenti attacchi convulsivi, un ritardo mentale chiaramente espresso o un significativo deficit visivo dovuto alla cataratta, tali pazienti sono già considerati disabili.
Diagnosi di pseudoipoparatiroidismo
I sintomi sono abbastanza specifici e se iniziano a comparire, è necessario contattare immediatamente un medico qualificato. È la diagnosi precoce e corretta che garantisce il successo del trattamento.
Poiché la malattia di Albright è congenita, di solito viene diagnosticata al paziente durante l'infanzia (età prescolare, scuola primaria). I medici genetici traggono conclusioni sulla base dei sintomi clinici; vengono utilizzati anche metodi diagnostici di laboratorio e strumentali.
In particolare, per chiarire la diagnosi, il paziente dona il sangue (nei pazienti con pseudoipoparatiroidismo, il livello di calcio è basso e il fosforo e l'ormone paratiroideo sono alti, l'attività della fosfatasi alcalina nel sangue è alta), l'urina (nel paziente, questo l'analisi mostrerà una diminuzione del fosforo e del calcio escreti). Vengono eseguiti anche esami speciali che mostrano la sensibilità del tessuto tubulare renale del paziente all’ormone paratiroideo. A volte diventa necessario valutare il livello di idrossiprolina.
I metodi strumentali prevedono la diagnostica a raggi X (è più istruttiva quando si identificano cambiamenti specifici nel tessuto osseo e muscolare).
Trattamento dello pseudoipoparatiroidismo
Questa rara patologia ereditaria viene trattata con integratori di calcio. Il medico seleziona la dose in base al contenuto di calcio necessario per mantenere l'omeostasi. E affinché il calcio venga assorbito senza problemi, nel regime terapeutico sono inclusi anche i preparati di vitamina D. Inoltre, il medico per un paziente con pseudoipoparatiroidismo svilupperà una dieta in cui la quantità di alimenti contenenti fluoro sarà limitata. Questa dieta aiuterà a normalizzare la concentrazione di calcio nel sangue e ad eliminare i segni di iperparatiroidismo secondario.
Se la malattia di Albright è accompagnata da altri disturbi endocrini, potrebbe essere necessaria una terapia ormonale sostitutiva. Le convulsioni vengono alleviate con una soluzione di cloruro di calcio o gluconato di calcio, somministrata per via endovenosa. Se appare una cataratta, viene trattata da un oftalmologo e uno psicologo aiuta ad alleviare il deterioramento cognitivo.
Con un regime terapeutico ben ponderato, la prognosi è molto favorevole. Ma tutti coloro che hanno avuto lo pseudoipoparatiroidismo dovrebbero consultare regolarmente i genetisti per prevenire la malattia nei loro discendenti.
Prevenzione dello pseudoipoparatiroidismo
Considerando che la malattia è ereditaria, l'unico modo per prevenirla per coloro che hanno una storia familiare di pseudoipoparatiroidismo è visitare regolarmente i genetisti medici per consultazioni.