Distrofia granulare. Distrofia corneale granulare. Cambiamenti distrofici associati a disturbi del metabolismo lipidico
La distrofia (dal greco dis - disordine e tropho - nutrire) è un cambiamento strutturale quantitativo e qualitativo nelle cellule e/o nella sostanza intercellulare di organi e tessuti, causato da una violazione dei processi metabolici. Nelle distrofie, a seguito di disturbi trofici, vari prodotti metabolici (proteine, grassi, carboidrati, minerali, acqua) si accumulano nelle cellule o nella sostanza intercellulare. L'essenza morfologica delle distrofie si esprime in:
- un aumento o una diminuzione della quantità di sostanze normalmente contenute nel corpo (ad esempio, un aumento della quantità di grasso nei depositi di grasso);
- cambiamenti di qualità, cioè proprietà fisico-chimiche delle sostanze normalmente inerenti al corpo (ad esempio, cambiamenti nelle proprietà tintoriali delle fibre di collagene con gonfiore mucoide e cambiamenti fibrinoidi);
- la comparsa di sostanze ordinarie in un luogo insolito (ad esempio, l'accumulo di vacuoli di grasso nel citoplasma delle cellule degli organi parenchimali durante la degenerazione grassa);
- la comparsa e l'accumulo di nuove sostanze che normalmente non sono presenti in esso (ad esempio la proteina amiloide).
Pertanto, la distrofia è un'espressione morfologica di disordini metabolici di cellule e tessuti.
Tra i meccanismi per il mantenimento del normale trofismo si distinguono quello cellulare ed extracellulare.
I meccanismi cellulari sono forniti dall'organizzazione strutturale della cellula e dalla sua autoregolazione, prevista dal codice genetico.
I meccanismi extracellulari del trofismo sono forniti dai sistemi di trasporto (sangue, linfa) e integrativi (nervoso, endocrino, umorale) della sua regolazione.
La causa diretta dello sviluppo delle distrofie può essere:
- Sostanze tossiche (comprese le tossine dei microrganismi).
- Agenti fisici e chimici: alte e basse temperature, alcuni prodotti chimici (acidi, alcali, sali di metalli pesanti, molte sostanze organiche), radiazioni ionizzanti.
- Fermentopatia acquisita o ereditaria (enzimopatia).
- Virus.
- Disturbi nella funzione dei sistemi energetici e di trasporto che assicurano il metabolismo e la conservazione strutturale dei tessuti (cellule), in cui si verifica quanto segue:
ü Ipoglicemia. Un basso livello di glucosio nel sangue (ipoglicemia) porta a una produzione insufficiente di molecole di adenosina trifosfato (ATP), che è più pronunciata nel cervello.
ü Ipossia: la mancanza di ossigeno nelle cellule (ipossia) può verificarsi quando:
- ostruzione delle vie aeree o malattia che impedisce l'ossigenazione del sangue nei polmoni;
- ischemia o interruzione del flusso sanguigno nei tessuti a causa di disturbi circolatori generali o locali;
- anemia (cioè con una diminuzione del livello di emoglobina nel sangue), che porta ad una diminuzione del trasporto di ossigeno da parte del sangue;
- alterazione della struttura dell'emoglobina (ad esempio in caso di avvelenamento da monossido di carbonio (CO)), che porta alla formazione di metaemoglobina, che non è in grado di trasportare ossigeno; questo porta allo stesso risultato dell'anemia.
- Disturbi della regolazione endocrina e nervosa:
- Malattie degli organi endocrini (tireotossicosi, diabete, iperparatiroidismo, ecc.)
- Malattie del sistema nervoso centrale e periferico (innervazione compromessa, tumori cerebrali).
Morfogenesi delle distrofie
Tra i meccanismi che portano allo sviluppo dei cambiamenti caratteristici delle distrofie ci sono l'infiltrazione, la decomposizione (fanerosi), la sintesi perversa e la trasformazione.
Infiltrazione - penetrazione eccessiva di prodotti metabolici dal sangue e dalla linfa nelle cellule o nella sostanza intercellulare e/o interruzione della loro inclusione nel metabolismo con conseguente accumulo. Ad esempio, l'infiltrazione della proteina epiteliale dei tubuli prossimali dei reni nella sindrome nefrosica, l'infiltrazione dell'intima dell'aorta e delle grandi arterie con lipoproteine nell'aterosclerosi.
Decomposizione (fanerosi) - decomposizione di sostanze chimicamente complesse. Ad esempio, la rottura dei complessi lipoproteici e l'accumulo di grasso libero nella cellula (degenerazione grassa dei cardiomiociti durante l'intossicazione da difterite). La rottura dei complessi polisaccaridi-proteine è alla base dei cambiamenti fibrinoidi nel tessuto connettivo nelle malattie reumatiche.
Trasformazione - la transizione da una sostanza all'altra. Questa è, ad esempio, la trasformazione dei carboidrati in grassi nel diabete mellito, una maggiore polimerizzazione del glucosio in glicogeno, ecc.
Sintesi perversa - è la sintesi nelle cellule o nei tessuti di sostanze che normalmente non si trovano in essi. Questi includono: la sintesi della proteina amiloide anormale nella cellula e la formazione di complessi anormali proteina amiloide-polisaccaride nella sostanza intercellulare, la sintesi della proteina ialina alcolica da parte degli epatociti, la sintesi del glicogeno nell'epitelio di uno stretto segmento del nefrone nel diabete mellito.
Classificazione.
A seconda della localizzazione dei disturbi metabolici:
ü parenchimale;
ü stroma-vascolare;
ü misto.
Secondo la predominanza delle violazioni dell'uno o dell'altro tipo di scambio:
ü proteine;
ü grasso;
ü carboidrati;
üminerale.
A seconda dell'influenza dei fattori genetici:
üacquistato;
ü ereditario.
Secondo la prevalenza del processo:
ülocale.
DISTROFIA PARANCHIMATA
Dai tempi di R. Virchow molti patologi hanno e continuano a classificare la cosiddetta “distrofia granulare”, che lo stesso R. Virchow definì “gonfiore torbido”, tra le distrofie proteiche parenchimali. È così che è consuetudine designare il processo in cui appare una granularità pronunciata nel citoplasma delle cellule degli organi parenchimali. Gli organi aumentano di dimensioni, diventano flaccidi e opachi quando vengono tagliati, come se fossero scottati dall'acqua bollente. Studi al microscopio elettronico e istoenzimatici della “distrofia granulare” hanno dimostrato che essa non si basa sull’accumulo di proteine nel citoplasma, ma sull’iperplasia (cioè un aumento del numero) delle ultrastrutture delle cellule degli organi parenchimali come espressione di la tensione funzionale di questi organi in risposta a varie influenze.
DISTROFIE PROTEICHE PARANCHIMATE
Attualmente, le distrofie proteiche parenchimali (disproteinosi) comprendono le goccioline ialine, quelle idropiche e quelle cornee. Tuttavia, va sottolineato che la distrofia cornea, secondo il meccanismo del suo sviluppo, non è correlata alle precedenti.
Distrofia delle goccioline ialine
Nella distrofia delle goccioline ialine, nel citoplasma compaiono grandi grumi e goccioline proteiche simili a ialine, che si fondono tra loro e riempiono il corpo cellulare. La base di questa distrofia è la coagulazione delle proteine citoplasmatiche con pronunciata distruzione degli elementi ultrastrutturali della cellula - necrosi coagulativa focale.
Questo tipo di disproteinosi si verifica spesso nei reni, meno spesso nel fegato e molto raramente nel miocardio.
L'aspetto degli organi con questa distrofia non ha caratteristiche caratteristiche. I cambiamenti macroscopici sono caratteristici di quelle malattie in cui si verifica la distrofia delle goccioline ialine.
Nei reni, all'esame microscopico, si trova l'accumulo di grandi grani di proteine rosa brillante - gocce ialine - nei nefrociti. In questo caso si osserva la distruzione dei mitocondri, del reticolo endoplasmatico e dell'orletto a spazzola. La base della distrofia delle goccioline ialine dei nefrociti è l'insufficienza dell'apparato vacuolare-lisosomiale dell'epitelio dei tubuli contorti prossimali e distali, che normalmente riassorbe le proteine. Pertanto questo tipo di distrofia nefrocitaria è molto comune nella sindrome nefrosica e riflette l'insufficienza di riassorbimento dei tubuli contorti rispetto alle proteine. Questa sindrome è una delle manifestazioni di molte malattie renali in cui è colpito principalmente il filtro glomerulare (glomerulonefrite, amiloidosi renale, nefropatia paraproteinemica, ecc.)
Nel fegato, all'esame microscopico, negli epatociti si trovano grumi e gocce di natura proteica: si tratta della ialina alcolica, che a livello ultrastrutturale rappresenta aggregati irregolari di microfibrille e inclusioni ialine di forma irregolare (corpi di Mallory). La formazione di questa proteina e dei corpi di Mallory è una manifestazione della funzione proteico-sintetica perversa dell'epatocita ed è costantemente rilevata nell'epatite alcolica.
L'esito della distrofia delle goccioline ialine è sfavorevole: termina con un processo irreversibile che porta alla necrosi coagulativa totale della cellula.
Il significato funzionale di questa distrofia è molto grande: c'è una forte diminuzione della funzione degli organi. La distrofia delle goccioline ialine dell'epitelio tubulare renale è associata alla comparsa di proteine (proteinuria) e cilindri (cilindruria) nelle urine, perdita di proteine plasmatiche (ipoproteinemia) e disturbi dell'equilibrio elettrolitico. La degenerazione delle goccioline ialine degli epatociti è spesso la base morfologica dei disturbi di molte funzioni epatiche.
Distrofia idropica
È caratterizzato dalla comparsa nella cellula di vacuoli pieni di fluido citoplasmatico. Il liquido si accumula nelle cisterne del reticolo endoplasmatico e nei mitocondri, meno spesso nel nucleo cellulare. Il meccanismo di sviluppo della distrofia idropica è complesso e riflette disturbi nel metabolismo dell'acqua, degli elettroliti e delle proteine, che portano a cambiamenti nella pressione colloido-osmotica nella cellula. Un ruolo importante è giocato dalla rottura della permeabilità delle membrane cellulari, accompagnata dalla loro disintegrazione. Ciò porta all'attivazione degli enzimi idrolitici lisosomiali, che rompono i legami intramolecolari con l'aggiunta di acqua. Essenzialmente, tali cambiamenti cellulari sono espressione di necrosi focale da liquefazione.
La distrofia idropica si osserva nell'epitelio della pelle e nei tubuli renali, negli epatociti, nelle cellule muscolari e nervose, nonché nelle cellule della corteccia surrenale.
Le ragioni per lo sviluppo della distrofia idropica in diversi organi sono ambigue. Nei reni si tratta di un danno al filtro glomerulare (glomerulonefrite, amiloidosi, diabete mellito), che porta all'iperfiltrazione e all'insufficienza del sistema enzimatico dei nefrociti, che normalmente garantisce il riassorbimento dell'acqua; avvelenamento da glicole, ipokaliemia. Nel fegato, la distrofia idropica si manifesta con epatite virale e tossica. Le cause della distrofia idropica dell'epidermide possono essere infezioni e allergie.
L'aspetto degli organi e dei tessuti cambia poco con la distrofia idropica.
Quadro microscopico: le cellule parenchimali sono aumentate di volume, il loro citoplasma è pieno di vacuoli contenenti liquido limpido. Il nucleo si sposta verso la periferia, talvolta vacuola o si restringe. Un aumento dell'idropia porta alla disintegrazione delle ultrastrutture cellulari e al trabocco della cellula con acqua, alla comparsa di palloncini pieni di liquido, pertanto tali cambiamenti sono chiamati distrofia del palloncino.
L'esito della distrofia idropica è solitamente sfavorevole; termina con la necrosi totale del colliquamento della cellula. Pertanto, la funzione degli organi e dei tessuti nella distrofia idropica è drasticamente ridotta.
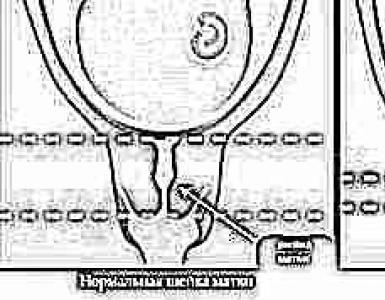
Distrofia cornea
La distrofia cornea, o cheratinizzazione patologica, è caratterizzata da un'eccessiva formazione di sostanza cornea nell'epitelio cheratinizzante (ipercheratosi, ittiosi) o dalla formazione di sostanza cornea dove normalmente non esiste - cheratinizzazione patologica sulle mucose, ad esempio nella cavità orale cavità (leucoplachia), esofago, cervice. La distrofia cornea può essere locale o generale, congenita o acquisita.
Le cause della distrofia cornea sono molteplici: infiammazione cronica associata ad agenti infettivi, azione di fattori fisici e chimici, carenze vitaminiche, disturbi congeniti dello sviluppo cutaneo, ecc.
L’esito può essere duplice: l’eliminazione della causa causale all’inizio del processo può portare al ripristino dei tessuti, ma nei casi avanzati si verifica la morte cellulare.
Il significato della distrofia cornea è determinato dal suo grado, prevalenza e durata. La cheratinizzazione patologica a lungo termine della mucosa (leucoplachia) può essere una fonte di sviluppo del cancro. L'ittiosi congenita grave è, di regola, incompatibile con la vita.
DISTROFIA GRASSA PARANCHIMATA (LIPIDOSI)
La degenerazione grassa parenchimale è una manifestazione strutturale di un disturbo del metabolismo dei lipidi citoplasmatici, che può esprimersi nell'accumulo di grasso allo stato libero in cellule dove non si trova normalmente.
Le cause della degenerazione grassa sono varie:
- carenza di ossigeno (ipossia tissutale), motivo per cui la degenerazione grassa è così comune nelle malattie del sistema cardiovascolare, nelle malattie polmonari croniche, nell'anemia, nell'alcolismo cronico, ecc. In condizioni di ipossia, le parti dell'organo che sono sotto tensione funzionale sono principalmente ricercato;
- infezioni gravi o a lungo termine (difterite, tubercolosi, sepsi);
- intossicazione (fosforo, arsenico, cloroformio, alcol), che porta a disordini metabolici;
- carenze vitaminiche e nutrizione unilaterale (proteine insufficienti), accompagnate da una carenza di enzimi e fattori lipotropici, necessari per il normale metabolismo dei grassi della cellula.
La degenerazione grassa parenchimale è caratterizzata principalmente dall'accumulo di trigliceridi nel citoplasma delle cellule parenchimali. Quando la connessione delle proteine con i lipidi viene interrotta - decomposizione, che avviene sotto l'influenza di infezioni, intossicazioni, prodotti di perossidazione lipidica - si verifica la distruzione delle strutture della membrana della cellula e nel citoplasma compaiono lipidi liberi, che sono il substrato morfologico del parenchima degenerazione grassa. Si osserva più spesso nel fegato, meno spesso nel rene e nel miocardio, ed è considerata una risposta non specifica a un gran numero di tipi di danno.
Segni microscopici di degenerazione grassa: qualsiasi grasso presente nei tessuti si dissolve nei solventi utilizzati per colorare i campioni di tessuto per l'esame microscopico. Pertanto, con il cablaggio convenzionale e la colorazione dei tessuti (colorazione con ematossilina ed eosina), le cellule nei primi stadi della degenerazione grassa presentano un citoplasma pallido e schiumoso. Quando le inclusioni grasse aumentano, nel citoplasma compaiono piccoli vacuoli.
La colorazione specifica del grasso richiede l'uso di sezioni congelate ottenute da tessuto fresco. Nelle sezioni congelate, il grasso rimane nel citoplasma, dopodiché le sezioni vengono colorate con coloranti speciali. Istochimicamente, i grassi vengono rilevati utilizzando diversi metodi: Sudan IV, rosso grasso O e bocca scarlatta li colorano di rosso, Sudan III - arancione, Sudan nero B e acido osmico - nero, il solfato blu del Nilo colora gli acidi grassi in blu scuro. e neutro grassi - in rosso. Utilizzando un microscopio polarizzatore è possibile distinguere tra lipidi isotropi e anisotropi. I lipidi anisotropi come il colesterolo e i suoi esteri mostrano una caratteristica birifrangenza.
La degenerazione del fegato grasso si manifesta con un forte aumento del contenuto e un cambiamento nella composizione dei grassi negli epatociti. Nelle cellule del fegato compaiono prima i granuli lipidici (obesità polverizzata), poi piccole goccioline (obesità da piccole goccioline), che successivamente si fondono in grandi goccioline (obesità da goccioline grandi) o in un vacuolo di grasso, che riempie l'intero citoplasma e spinge il nucleo verso la periferia. Le cellule del fegato modificate in questo modo assomigliano alle cellule adipose. Più spesso, la deposizione di grasso nel fegato inizia alla periferia, meno spesso - al centro dei lobuli; con distrofia significativamente pronunciata, l'obesità delle cellule epatiche è diffusa.
Macroscopicamente il fegato in degenerazione grassa si presenta ingrossato, anemico, di consistenza pastosa, di colore giallo o giallo ocra, con riflessi untuosi al taglio. Durante il taglio è visibile uno strato di grasso sulla lama del coltello e sulla superficie tagliata.
Tipi di fegato grasso:
Il fegato grasso acuto è una condizione rara ma grave associata a danno epatico acuto. Nel fegato grasso acuto, i trigliceridi si accumulano nel citoplasma sotto forma di piccoli vacuoli delimitati dalla membrana (piccolo fegato grasso).
Il fegato grasso cronico può verificarsi con alcolismo cronico, malnutrizione e avvelenamento da alcune epatotossine. Le goccioline di grasso nel citoplasma si combinano per formare grandi vacuoli (malattia del fegato grasso da goccioline grandi). La localizzazione dei cambiamenti grassi nel lobulo epatico dipende dalle ragioni che li hanno causati. Anche nel fegato grasso cronico grave, raramente vi è evidenza clinica di disfunzione epatica.
La degenerazione grassa del miocardio è caratterizzata dall'accumulo di trigliceridi nel miocardio.
Cause di degenerazione grassa del miocardio:
condizioni ipossiche croniche, soprattutto con anemia grave. Nella degenerazione grassa cronica, le strisce gialle si alternano ad aree rosso-marroni (“cuore di tigre”). I segni clinici di solito non sono molto pronunciati.
il danno tossico, come la miocardite difterica, provoca una degenerazione grassa acuta. Macroscopicamente il cuore è flaccido, è presente una colorazione gialla diffusa, il cuore appare ingrandito di volume, le sue camere sono distese; il quadro clinico mostra segni di insufficienza cardiaca acuta.
La degenerazione grassa del miocardio è considerata l'equivalente morfologico del suo scompenso. La maggior parte dei mitocondri si disintegrano e le striature trasversali delle fibre scompaiono. Lo sviluppo della degenerazione grassa del miocardio è spesso associato non alla distruzione dei complessi della membrana cellulare, ma alla distruzione dei mitocondri, che porta all'interruzione dell'ossidazione degli acidi grassi nella cellula. Nel miocardio, la degenerazione grassa è caratterizzata dalla comparsa di minuscole goccioline di grasso nelle cellule muscolari (obesità polverizzata). Con l'aumento dei cambiamenti, queste goccioline (obesità da piccole goccioline) sostituiscono completamente il citoplasma. Il processo è di natura focale e si osserva in gruppi di cellule muscolari situati lungo il ginocchio venoso dei capillari e delle piccole vene, il più delle volte subendo- e subepicardiche.
Nei reni, con la degenerazione grassa, i grassi compaiono nell'epitelio dei tubuli prossimali e distali. Di solito si tratta di grassi neutri, fosfolipidi o colesterolo, che si trovano non solo nell'epitelio tubolare, ma anche nello stroma. I grassi neutri nell'epitelio del segmento stretto e nei dotti collettori si verificano come fenomeno fisiologico.
Aspetto dei reni: sono ingrossati, flaccidi (densi se associati ad amiloidosi), la corteccia è gonfia, grigia con macchioline gialle, evidenti sulla superficie e sulla sezione.
Il meccanismo di sviluppo della degenerazione renale grassa è associato all'infiltrazione di grasso nell'epitelio dei tubuli renali durante la lipemia e l'ipercolesterolemia (sindrome nefrosica), che porta alla morte dei nefrociti.
L'esito della degenerazione grassa dipende dal suo grado. Se non è accompagnato da una grave disgregazione delle strutture cellulari, di norma risulta reversibile. Una profonda interruzione del metabolismo lipidico cellulare nella maggior parte dei casi termina con la morte cellulare.
Il significato funzionale della degenerazione grassa è grande: il funzionamento degli organi viene bruscamente interrotto e in alcuni casi si interrompe. Alcuni autori hanno espresso l'idea che il grasso compaia nelle cellule durante il periodo di convalescenza e l'inizio della riparazione. Ciò è coerente con le idee biochimiche sul ruolo della via del pentoso fosfato per l’utilizzo del glucosio nei processi anabolici, che è anche accompagnato dalla sintesi dei grassi.
DISTROFIE DA CARBOIDRATI PARANCHIMATE
I carboidrati, che sono determinati nelle cellule e nei tessuti e possono essere identificati istochimicamente, sono suddivisi in polisaccaridi, di cui nei tessuti animali vengono rilevati solo glicogeno, glicosaminoglicani (mucopolisaccaridi) e glicoproteine. Tra i glicosaminoglicani ci sono quelli neutri, strettamente legati alle proteine, e quelli acidi, che comprendono l'acido ialuronico, l'acido condroitinsolforico e l'eparina. I glicosaminoglicani acidi, come biopolimeri, sono in grado di formare composti deboli con un numero di metaboliti e di trasportarli. I principali rappresentanti delle glicoproteine sono mucine e mucoidi. Le mucine costituiscono la base del muco prodotto dall'epitelio delle mucose e delle ghiandole; i mucoidi fanno parte di molti tessuti.
Metodi istochimici per l'identificazione dei carboidrati. Polisaccaridi, glicosaminoglicani e glicoproteine vengono rilevati dalla reazione PAS. L'essenza della reazione è che dopo l'ossidazione con acido periodico (o reazione con periodato di potassio), le aldeidi risultanti danno un colore rosso con la fucsina di Schiff. Per rilevare il glicogeno, la reazione PHIK è integrata dal controllo enzimatico - trattamento delle sezioni con amilasi. Il glicogeno è colorato di rosso dal carminio di Best. I glicosaminoglicani e le glicoproteine vengono determinati utilizzando diversi metodi, di cui i più comunemente utilizzati sono le colorazioni con blu di toluidina o blu di metilene. Queste colorazioni permettono di identificare le sostanze cromotropiche che danno origine alla reazione di metacromasia. Il trattamento di sezioni di tessuto con ialuronidasi (batterica, testicolare) seguito dalla colorazione con gli stessi coloranti consente di differenziare diversi glicosaminoglicani; ciò è possibile anche modificando il pH del colorante.
La distrofia parenchimale dei carboidrati può essere associata a un alterato metabolismo del glicogeno o delle glicoproteine.
Disturbo del metabolismo del glicogeno
Nel diabete mellito, il cui sviluppo è associato alla patologia delle cellule β delle isole pancreatiche, che causa un'insufficiente produzione di insulina, vi è un uso insufficiente del glucosio da parte dei tessuti, un aumento del suo contenuto nel sangue (iperglicemia) e l'escrezione nelle urine (glicosuria). Le riserve di glicogeno tissutale diminuiscono drasticamente. Ciò riguarda principalmente il fegato, in cui la sintesi del glicogeno viene interrotta, il che porta alla sua infiltrazione con grassi: si sviluppa la degenerazione del fegato grasso; allo stesso tempo, nei nuclei degli epatociti compaiono inclusioni di glicogeno, che diventano leggeri (nuclei "vuoti").
La glicosuria è associata ad alterazioni renali caratteristiche del diabete. Sono espressi nell'infiltrazione di glicogeno dell'epitelio tubulare, principalmente nei segmenti stretti e distali. L'epitelio diventa alto, con citoplasma leggermente schiumoso; nel lume dei tubuli sono visibili anche granuli di glicogeno. Questi cambiamenti riflettono lo stato della sintesi del glicogeno (polimerizzazione del glucosio) nell'epitelio tubulare durante il riassorbimento dell'ultrafiltrato plasmatico ricco di glucosio.
Nel diabete non vengono colpiti solo i tubuli renali, ma anche i glomeruli e le loro anse capillari, la cui membrana basale diventa permeabile agli zuccheri e alle proteine plasmatiche. Si verifica una delle manifestazioni della microangiopatia diabetica: la glomerulosclerosi intercapillare (diabetica).
Le distrofie ereditarie dei carboidrati, che si basano su disturbi del metabolismo del glicogeno, sono chiamate glicogenosi. La glicogenosi è causata dall'assenza o dal deficit dell'enzima coinvolto nella degradazione del glicogeno immagazzinato e appartiene quindi alle enzimopatie ereditarie o malattie da accumulo. Attualmente sono stati ben studiati 6 tipi di glicogenosi, causati dalla carenza ereditaria di 6 diversi enzimi. Si tratta delle malattie di Gierke (tipo I), di Pompe (tipo II), di McArdle (tipo V) e di Hers (tipo VI), in cui la struttura del glicogeno accumulato nei tessuti non è disturbata, e di Forbes-Cory (tipo III) e Malattie di Andersen (tipo IV), in cui è bruscamente cambiato.
La diagnosi morfologica della glicogenosi di un tipo o dell'altro è possibile esaminando una biopsia utilizzando metodi istoenzimatici, nonché tenendo conto della localizzazione del glicogeno accumulato.
Distrofie dei carboidrati associate ad alterato metabolismo delle glicoproteine
Quando il metabolismo delle glicoproteine nelle cellule o nella sostanza intercellulare viene interrotto, si accumulano mucine e mucoidi, chiamati anche sostanze mucose o simili al muco. A questo proposito, quando il metabolismo delle glicoproteine viene interrotto, si parla di distrofia delle mucose.
Esame microscopico. Permette di rilevare non solo un aumento della formazione di muco, ma anche cambiamenti nelle proprietà fisico-chimiche del muco. Molte cellule secernenti muoiono e desquamano, i dotti escretori delle ghiandole vengono ostruiti dal muco, il che porta allo sviluppo di cisti. Spesso in questi casi si associa un'infiammazione. Il muco può chiudere i lumi dei bronchi, provocando la comparsa di atelettasia e focolai di polmonite.
A volte non è vero muco che si accumula nelle strutture ghiandolari, ma sostanze simili al muco (pseudomucine). Queste sostanze possono diventare più dense e assumere il carattere di un colloide. Poi parlano della distrofia colloidale, che si osserva, ad esempio, nel gozzo colloidale.
Le cause della distrofia mucosa sono varie, ma molto spesso si tratta di un'infiammazione delle mucose a seguito dell'azione di vari agenti irritanti patogeni (infiammazione catarrale).
La distrofia della mucosa è alla base di una malattia sistemica ereditaria chiamata fibrosi cistica, caratterizzata da un cambiamento nella qualità del muco secreto dall'epitelio delle ghiandole mucose: il muco diventa denso e viscoso, viene scarsamente escreto, il che provoca lo sviluppo di cisti da ritenzione e sclerosi (fibrosi cistica). Sono interessati l'apparato esocrino del pancreas, le ghiandole dell'albero bronchiale, il tratto digerente e urinario, i dotti biliari, le ghiandole sudoripare e lacrimali.
Il risultato è in gran parte determinato dal grado e dalla durata della produzione di muco in eccesso. In alcuni casi, la rigenerazione dell'epitelio porta al completo ripristino della mucosa, in altri si atrofizza e successivamente diventa sclerotica, il che influisce naturalmente sulla funzione dell'organo.
Definizione
Distrofia granulareè una distrofia parenchimale, caratterizzata dalla comparsa nel citoplasma di cellule granulari, che sono mitocondri rigonfi. Considerato come un tipo di distrofia proteica.
Evento.
Più spesso osservato negli epatociti, nefrociti, cardiomiociti. Il fenomeno è estremamente comune, poiché si manifesta principalmente durante l'ipossia circolatoria, che si osserva in molte condizioni patologiche.
Condizioni di accadimento.
- Caduta della pressione arteriosa sistemica accompagnata da ipoperfusione tissutale.
- Insufficienza relativa (inadeguatezza) dell'apporto di sangue a un organo in condizioni di intenso funzionamento.
- Gonfiore dei tessuti, accompagnato da una ridotta diffusione dell'ossigeno nella cellula.
Meccanismi di accadimento.
In condizioni ipossiche, la fosforilazione ossidativa e la sintesi di ATP diminuiscono drasticamente. A causa della carenza di energia, il lavoro della pompa ionica - K + /Na + - ATPasi - incorporata nelle membrane degli organelli e nella membrana cellulare e che garantisce la rimozione attiva degli ioni Na + all'esterno della cellula, viene interrotto. Negli organelli, principalmente nei mitocondri, si verifica l'accumulo di questi ioni e, di conseguenza, dell'acqua. I mitocondri gonfi assumono l'aspetto di grani, visibili al microscopio ottico.
Immagine macroscopica.
I cambiamenti negli organi interni in questo tipo di distrofia sono descritti come “gonfiore torbido”. Nei reni e nel fegato, che hanno una capsula, il tessuto sporge leggermente oltre i bordi del taglio. Il rigonfiamento è associato alla stessa disfunzione delle pompe ioniche e all'accumulo di quantità eccessive di acqua nella cellula. La lucentezza torbida e l'aspetto opaco di un organo su una sezione è probabilmente dovuto al fatto che lo strato superficiale della sezione risulta otticamente più denso a causa dei mitocondri gonfi e riflette peggio la luce che cade su di esso, che noi percepiamo.
Immagine microscopica.
Nel citoplasma delle cellule, ad alto ingrandimento, si rivelano piccoli grani che hanno un colore rosa intenso quando colorati con eosina. Sono particolarmente evidenti quando il microcampione è colorato con azzurro ed eosina. A livello ultrastrutturale i mitocondri sono ingranditi di 2-5 volte o più con matrice illuminata, con creste frammentate, spesso poco visibili. Lo strato esterno della membrana a doppio strato è assente in numerosi organelli. In alcuni casi, i cambiamenti nei mitocondri sono accompagnati dall'espansione delle cisterne del reticolo endoplasmatico rugoso.
Significato clinico.
Il fatto stesso della distrofia granulare indica una carenza di ossigeno ed energia nella cellula, che ha un effetto estremamente sfavorevole sulla sua funzione, soprattutto quando si tratta di cardiomiociti. D'altra parte, il rigonfiamento dei mitocondri e la frammentazione delle creste al loro interno interrompono ulteriormente la sintesi di ATP. Anche la “danza dei mitocondri” normalmente esistente viene bruscamente interrotta: i mitocondri che sono diventati “goffi” garantiscono scarsamente la consegna di ATP agli organelli che consumano energia, che non possono che influenzare negativamente il funzionamento dell'intera cellula.
La distrofia granulare può manifestarsi in pochi minuti e scomparire rapidamente quando viene ripristinato il normale regime di ossigeno nella cellula: le pompe ioniche pompano sia l'eccesso di Na 5 + che l'acqua dai mitocondri, i mitocondri si gonfiano e quelli il cui strato esterno della membrana viene distrutto, viene catturato dai lisosomi e utilizzato.
A seconda delle strutture in cui sono prevalentemente localizzati i cambiamenti distrofici, questi si dividono in parenchimali, stroma-vascolari (mesenchimali) e misti.
A seconda della predominanza dei disturbi dell'uno o dell'altro tipo di metabolismo, si distinguono le distrofie proteiche, grasse, carboidratiche, minerali e miste.
Si distinguono anche le distrofie acquisite ed ereditarie e, a seconda della prevalenza, generale e locale.
Distrofie parenchimali
In queste distrofie, le sostanze si accumulano nelle cellule del parenchima di vari organi, come miocardiociti, epatociti, neuroni, cellule dei tubuli renali e ghiandole surrenali. In base al tipo di disturbo metabolico, queste distrofie si dividono in proteiche (disproteinosi), grassi (lipidosi) e carboidrati.
Le distrofie proteiche parenchimali (disproteinosi) sono caratterizzate da un metabolismo alterato delle proteine citoplasmatiche in uno stato libero o legato. Questi includono la distrofia granulare, idropica, a goccia ialina e cornea.
Distrofia granulare.
Le cause della distrofia granulare possono essere disturbi circolatori, infezioni, intossicazioni e altri fattori che portano ad una diminuzione dell'intensità dei processi redox della cellula. In questo processo, gli organi risultano ingrossati, flaccidi, opachi e opachi quando vengono tagliati. Ciò dà motivo di chiamare la distrofia granulare un gonfiore torbido (opaco) degli organi.
Microscopicamente le cellule sono ingrandite, il loro citoplasma è torbido e ricco di granuli proteici. La microscopia elettronica nella distrofia granulare mostra un aumento del numero (iperplasia) e un rigonfiamento degli organelli cellulari, che otticamente assomigliano a granuli proteici. Tale iperplasia delle ultrastrutture cellulari è attualmente considerata come una pronunciata tensione funzionale degli organi a vari influssi.
L'esito della distrofia granulare è diverso. Nella maggior parte dei casi è reversibile, ma se non si eliminano le cause che l'hanno provocata, può trasformarsi in degenerazione gocciolina ialina, idropica o grassa.
DISTROFIA GRANULARE
distrofia granulare, gonfiore torbido, il tipo più comune di distrofia proteica, caratterizzata da una violazione delle proprietà colloidali del citoplasma delle cellule con l'identificazione di proteine in esso contenute sotto forma di grani. Cause Z.d.: infezioni, alimentazione inadeguata, intossicazioni, disturbi della circolazione sanguigna e linfatica; fattori che causano ipossia tissutale, acidosi o, meno comunemente, alcalosi. Z.d. più pronunciato nel fegato, nei reni, nel miocardio e anche nei muscoli scheletrici. Inizialmente, si manifesta con gonfiore, annebbiamento del citoplasma e quindi comparsa di granularità in esso. Essenza Z.d. consiste in un cambiamento del metabolismo proteico nella cellula, in fermentopatia con conseguente danno ai suoi organelli. Gli organi sono ingrossati, flaccidi, anemici, la loro superficie tagliata è opaca, di colore grigiastro, il tessuto muscolare ricorda la carne scottata con acqua bollente. Le funzioni degli organi colpiti sono compromesse. Z.d.è un processo reversibile, ma nei casi più gravi può svilupparsi in degenerazione e necrosi idropica, a goccia ialina o grassa. Z.d. deve essere distinto dalla sintesi proteica fisiologica nella cellula con accumulo di granuli proteici e dalle alterazioni post mortem negli organi (ottusità cadaverica). Vedi anche la letteratura sotto questo articolo.
Dizionario enciclopedico veterinario. - M.: "Enciclopedia sovietica". Il redattore capo V.P. Shishkov. 1981 .
Scopri cos'è la “DISTROFIA GRANULARE” in altri dizionari:
Distrofia di cellule e tessuti- una violazione del metabolismo tissutale o cellulare, accompagnata da alcuni cambiamenti strutturali nelle cellule e nella sostanza intercellulare. Lo sviluppo della distrofia si basa su disturbi dei meccanismi regolatori del trofismo congenito o acquisito... ... Enciclopedia medica
- (dal prefisso greco dis che indica un disturbo e trophē nutrizione), cambiamenti qualitativi nella composizione chimica, proprietà fisico-chimiche, struttura e funzioni dei tessuti associati a disturbi metabolici; il processo dietro...
La distrofia epatica dei pesci è un nome collettivo che unisce una varietà di processi patologici che si verificano nel fegato dei pesci a causa di disturbi metabolici. Le degenerazioni più comunemente osservate sono quella granulare (proteica), idropica e grassa.… … Dizionario enciclopedico veterinario
distrofia granulare- (d. granulosa; sinonimo rigonfiamento torbido) un tipo di parenchima proteico D., caratterizzato da rigonfiamento delle cellule con aspetto di granelli e goccioline di natura proteica nel citoplasma; l'organo diventa flaccido e la superficie tagliata diventa opaca... Ampio dizionario medico
Distrofia proteica parenchimale- manifestato dalla comparsa nel citoplasma di un gran numero di granuli di natura proteica. Si verifica nel fegato, nei reni, nel cuore, durante l'infezione e l'intossicazione. La distrofia è reversibile dopo la cessazione dell'azione del fattore traumatico. Indice 1 Ialino ... ... Wikipedia
Distrofia proteica- (disproteinosi) malattie associate a disturbi del metabolismo proteico. Si riferisce a uno dei tre tipi di distrofia (distrofia parenchimale). Le proteine sono uno dei componenti principali delle cellule e dei tessuti. La sua massa è di circa il 45%... Wikipedia
Disproteinosi parenchimali- processi dismetabolici (degenerativi, distrofici) con un disturbo predominante del metabolismo proteico, che si sviluppa principalmente nelle cellule parenchimali degli organi. Contenuti 1 Classificazione 2 Distrofia granulare ... Wikipedia
Cuore- I Cuore Il cuore (latino cor, greco cardia) è un organo fibromuscolare cavo che, funzionando come una pompa, assicura il movimento del sangue nel sistema circolatorio. Anatomia Il cuore è situato nel mediastino anteriore (mediastino) nel pericardio tra... ... Enciclopedia medica
RAME- vedi RAME (Cu) è contenuto nelle acque reflue provenienti da impianti di lavorazione del minerale, imprese metallurgiche, di ingegneria meccanica ed elettriche. Solfato di rame, carbonato, ossicloruro e arseniato di rame sono usati come alghicidi, fungicidi e molluschicidi. Rame… … Malattie dei pesci: una guida
DERIVATI SIMM-TRIAZINICI- vedi DERIVATI DELLE TRIAZINE SIMM (atrazina, prometrina, propazina, simazina, semeron) sono usati come agenti di controllo delle infestanti per colture in campo, e l'atrazina è raccomandata per la distruzione della vegetazione acquatica nel drenaggio dei collettori e nell'irrigazione... ... Malattie dei pesci: una guida
Questo tipo di distrofia è anche chiamata distrofia parenchimale o gonfiore torbido. Ciascuno di questi termini definisce una diversa essenza del processo patologico.
Si chiama distrofia parenchimale perché questo processo degenerativo si sviluppa negli organi parenchimali (reni, fegato, cuore); gonfiore torbido - perché le cellule del parenchima colpite prima si gonfiano, perdono i loro contorni e nel protoplasma appare una granularità simile a polvere, conferendogli un aspetto torbido. Il protoplasma delle cellule colpite si colora con coloranti acidi più del solito. I fori rigonfi, aumentando di volume, comprimono i capillari, provocando il pallore degli organi degenerati.
Gli organi macroscopicamente modificati aumentano di volume, si gonfiano, il loro disegno caratteristico è levigato, il tessuto ha un aspetto opaco, una tinta grigiastra.
I muscoli cardiaci affetti da distrofia parenchimale assomigliano a carne scottata con acqua bollente. Con la degenerazione granulare, i reni si gonfiano, hanno un colore pallido al taglio, i bordi sono leggermente arrotondati, la capsula viene rimossa meglio del solito, il confine tra gli strati corticale e midollo viene levigato.
La base della distrofia granulare è la disorganizzazione fisico-chimica del protoplasma cellulare e una diminuzione della dispersione dei suoi colloidi, che porta a disturbi funzionali dell'organo interessato.
Nei preparati istologici preparati nel modo consueto e colorati con ematossilina-eosina, la granularità caratteristica di questo processo non è sempre ben espressa. È meglio visibile quando il tessuto viene esaminato non tinto. A questo scopo, un pezzo del tessuto da esaminare viene diviso con aghi su un vetrino. Una goccia di soluzione salina viene versata sopra e coperta con un coprioggetto. Con un ingrandimento medio del microscopio, la granularità del protoplasma in questi casi è chiaramente visibile. Se una goccia di acido acetico diluito viene posta sotto un vetrino coprioggetto, i granuli proteici si gonfiano, il protoplasma si omogeneizza e diventa trasparente. I nuclei precedentemente non rilevabili diventano evidenti.
Istomedicina. Distrofia renale parenchimale( )
A basso ingrandimento del microscopio, i tubuli urinari contorti, tagliati obliquamente, si presentano sotto forma di ovali di varie dimensioni, e quelli tagliati trasversalmente hanno la forma di cerchi. I lumi dei tubuli sono ristretti in alcuni punti e riempiti con una massa proteica granulare; le cellule epiteliali sono colorate in modo diseguale,
Con un ingrandimento medio del microscopio, si nota che le cellule epiteliali dei tubuli contorti dei reni sono torbide, gonfie e in molti di essi i nuclei non sono visibili. Il protoplasma è a grana fine. I lumi dei tubuli sono parzialmente riempiti di detriti proteici.