Le principali cause dell’anemia emolitica sono: Anemia emolitica causata da agenti infettivi. Anemia emolitica acuta o indotta da farmaci
Soggetto: Anemia emolitica - congenita e acquisita .
Scopo dello studio: introdurre gli studenti al concetto di anemia emolitica, considerare varie varianti cliniche dell'anemia emolitica, diagnosi, diagnosi differenziale, complicanze. Studiare i cambiamenti nel quadro ematico in varie varianti cliniche dell'anemia emolitica.
Parole chiave:
Anemia emolitica;
Emolisi;
Microsferocitosi;
Membranopatia e fermentopatia;
Talassemia;
Anemia falciforme;
Crisi emolitica
Piano di studio dell'argomento:
Il concetto di anemia emolitica;
Classificazione delle anemie emolitiche ereditarie;
Membranopatie;
Malattia di Minkowski-Choffard;
Enzimopatie;
Anemia associata a deficit di G-6-PD nei globuli rossi;
Emoglobinopatie;
Talassemia;
Anemia falciforme;
Classificazione delle anemie emolitiche acquisite;
Principi generali di diagnosi e trattamento dell'anemia emolitica.
Presentazione del materiale didattico:
Si chiama anemia, in cui il processo di distruzione dei globuli rossi prevale sul processo di rigenerazione emolitico.
La morte naturale di un eritrocita (erythrodierez) avviene 90-120 giorni dopo la sua nascita negli spazi vascolari del sistema reticoloistiocitario, principalmente nei sinusoidi della milza e molto meno spesso direttamente nel flusso sanguigno. Con l'anemia emolitica si osserva la distruzione prematura (emolisi) dei globuli rossi. La resistenza dell'eritrocita alle varie influenze dell'ambiente interno è dovuta sia alle proteine strutturali della membrana cellulare (spettrina, anchirina, proteina 4.1, ecc.) Che alla sua composizione enzimatica, inoltre, all'emoglobina normale e alle proprietà fisiologiche del sangue e altri ambienti in cui circola l'eritrocito. Quando le proprietà di un eritrocita vengono alterate o il suo ambiente cambia, viene distrutto prematuramente nel flusso sanguigno o nel sistema reticoloistiocitario di vari organi, principalmente la milza.
Classificazione delle anemie emolitiche
Di solito si distinguono le anemie emolitiche ereditarie e acquisite, poiché hanno diversi meccanismi di sviluppo e differiscono nell'approccio al trattamento. Meno comunemente, le anemie emolitiche sono classificate in base alla presenza o assenza di immunopatologia, distinguendo tra anemie emolitiche autoimmuni e non immuni, che comprendono anemie emolitiche congenite, anemie emolitiche acquisite in pazienti con cirrosi epatica, nonché in presenza di valvole cardiache protesiche e la cosiddetta emoglobinuria di marcia.
Anemia emolitica Hanno una serie di caratteristiche che le distinguono dalle anemie di altra origine. Innanzitutto si tratta di anemie iperrigenerative, che si manifestano con ittero emolitico e splenomegalia. L'elevata reticolocitosi nell'anemia emolitica è dovuta al fatto che durante la disgregazione degli eritrociti si formano tutti gli elementi necessari per la costruzione di un nuovo eritrocita e, di regola, non vi è carenza di eritropoietina, vitamina B12, acido folico e ferro. La distruzione dei globuli rossi è accompagnata da un aumento del contenuto di bilirubina libera nel sangue; quando il suo livello supera i 25 µmol/l, compare l'isteria della sclera e della pelle. L'ingrossamento della milza (splenomegalia) è il risultato dell'iperplasia del tessuto reticoloistiocitario, causata dall'aumento dell'emolisi dei globuli rossi. Non esiste una classificazione generalmente accettata dell’anemia emolitica.
Anemia emolitica ereditaria.
UN. Membranopatie a causa della rottura della struttura della membrana eritrocitaria:
Violazione delle proteine della membrana degli eritrociti: microsferocitosi; ellissocitosi; stomatocitosi; piropoichilocitosi.
Disturbi dei lipidi della membrana eritrocitaria: acantocitosi, deficit dell'attività della lecitina-colesterolo aciltransferasi (LCAT), aumento del contenuto di lecitina nella membrana eritrocitaria, picnocitosi infantile.
B. Enzimepatie:
Carenza degli enzimi del ciclo del pentoso fosfato.
Carenza di attività degli enzimi glicolitici.
Carenza di attività degli enzimi del metabolismo del glutatione.
Carenza nell'attività degli enzimi coinvolti nell'utilizzo dell'ATP.
Carenza di attività della ribofosfato pirofosfato chinasi.
Attività compromessa degli enzimi coinvolti nella sintesi delle porfirine.
IN. Emoglobinopatie:
Causato da un'anomalia nella struttura primaria dell'emoglobina
Causato da una diminuzione della sintesi delle catene polipeptidiche che compongono l'emoglobina normale
Causato da un doppio stato eterozigote
Anomalie dell'emoglobina non accompagnate dallo sviluppo della malattia
Anemia emolitica acquisita
UN. Anemie emolitiche immunitarie:
Anemie emolitiche associate ad esposizione ad anticorpi: isoimmuni, eteroimmuni, transimmuni.
Anemia emolitica autoimmune: con agglutinine calde incomplete, con emolisina calda, con agglutinine fredde complete, associata ad emolisina fredda bifasica.
Anemia emolitica autoimmune con anticorpi contro l'antigene normocitario del midollo osseo.
B. Anemia emolitica associata ad alterazioni della membrana causate da mutazione somatica: EPN.
B. Anemia emolitica associata a danno meccanico alla membrana eritrocitaria.
D. Anemia emolitica associata a danno chimico ai globuli rossi (piombo, acidi, veleni, alcol).
D. Anemia emolitica dovuta a carenza di vitamine E e A.
L'anemia emolitica è un gruppo di malattie che differiscono per natura, quadro clinico e principi di trattamento, ma sono accomunate da un'unica caratteristica: l'emolisi dei globuli rossi. Tra le malattie del sangue, le anemie emolitiche rappresentano il 5% e tra tutte le anemie, le anemie emolitiche rappresentano l'11%. Il sintomo principale delle condizioni emolitiche è l'emolisi: una diminuzione dell'aspettativa di vita dei globuli rossi e la loro maggiore degradazione.
EZIOLOGIA E PATOGENESI. La norma fisiologica per la durata della vita dei globuli rossi varia da 100 a 120 giorni. Il globulo rosso ha un metabolismo potente e trasporta un carico funzionale colossale. Fornire le funzioni degli eritrociti è determinato dalla conservazione della struttura e della forma delle cellule e dei processi che assicurano il metabolismo dell'emoglobina. L'attività funzionale è assicurata dal processo di glicolisi, che porta alla sintesi di ATP, che fornisce energia ai globuli rossi. L'integrità strutturale e il normale metabolismo dell'emoglobina sono assicurati dalla proteina strutturale tripeptide glutatione. La forma è mantenuta dalle lipoproteine nella membrana degli eritrociti. Una proprietà importante dei globuli rossi è la loro capacità di deformarsi, che garantisce il libero passaggio dei globuli rossi quando entrano nei microcapillari e quando lasciano i seni della milza. La deformabilità dei globuli rossi dipende da fattori interni ed esterni. Fattori interni: viscosità (fornita dalla normale concentrazione di emoglobina nella parte centrale dell'eritrocita) e pressione oncotica all'interno dell'eritrocita (a seconda della pressione oncotica del plasma sanguigno, della presenza di cationi magnesio e potassio nell'eritrocita). Con un'elevata pressione oncotica del plasma, i suoi elementi si riversano nell'eritrocita, si deforma e scoppia. Il contenuto normale di magnesio e potassio dipende dal funzionamento del meccanismo di trasporto della membrana, che, a sua volta, dipende dal corretto rapporto tra componenti proteici e fosfolipidi nella membrana, ad es. se qualsiasi parte del programma genetico dell'eritrocita viene interrotta ( sintesi delle proteine di trasporto o di membrana), allora l’equilibrio è disturbato fattori interni, che porta alla morte del globulo rosso.
Con lo sviluppo dell'anemia emolitica, la durata della vita dei globuli rossi si riduce a 12-14 giorni. L'emolisi patologica è divisa in intravascolare e intracellulare. L'emolisi intravascolare è caratterizzata da un aumento dell'apporto di emoglobina nel plasma e dall'escrezione nelle urine sotto forma di emosiderina o immodificata. L'emolisi intracellulare è caratterizzata dalla rottura dei globuli rossi nel sistema reticolocitario della milza, che è accompagnata da un aumento del contenuto della frazione di bilirubina libera nel siero del sangue, dall'escrezione di urobilina nelle feci e nelle urine e da una tendenza alla colelitiasi e alla coledocolitiasi.
Malattia di Minkowski-Choffard (microsferocitosi ereditaria).
La malattia di Minkowski-Choffard è una malattia ereditaria, trasmessa con modalità autosomica dominante.
EZIOLOGIA E PATOGENESI. In pratica, un caso su quattro non viene ereditato. Ovviamente, questo tipo si basa su qualche mutazione spontanea, formata a seguito dell'azione di fattori teratogeni. Un difetto geneticamente ereditato nella proteina della membrana eritrocitaria porta ad un eccesso di ioni sodio e molecole d'acqua negli eritrociti, con conseguente formazione di forme patologiche di eritrociti di forma sferica (sferociti). A differenza dei normali globuli rossi biconcavi, non sono in grado di deformarsi quando passano attraverso gli stretti vasi dei seni splenici. Di conseguenza, il progresso nei seni della milza rallenta, alcuni globuli rossi vengono separati e si formano piccole cellule: microsferociti, che vengono rapidamente distrutti. I detriti dei globuli rossi vengono catturati dai macrofagi splenici, il che porta allo sviluppo della splenomegalia. L'aumento dell'escrezione di bilirubina con la bile provoca lo sviluppo di pleiocromia e colelitiasi. Come risultato della maggiore degradazione dei globuli rossi, aumenta la quantità di frazione libera di bilirubina nel siero del sangue, che viene escreta dall'intestino con le feci sotto forma di stercobilina e parzialmente nelle urine. Nella malattia di Minkovsky-Shoffard, la quantità di stercobilina rilasciata supera i livelli normali di 15-20 volte.
QUADRO ANATOMICO-PATOLOGICO. A causa del germogliamento eritroide, il midollo osseo nelle ossa tubolari e piatte è iperplastico e si nota eritrofagocitosi. Nella milza si osserva una diminuzione del numero e delle dimensioni dei follicoli, iperplasia dell'endotelio del seno e pronunciato riempimento di sangue della polpa. L'emosiderosi può essere rilevata nei linfonodi, nel midollo osseo e nel fegato.
CLINICA. Nel corso della malattia si alternano periodi di remissione e di riacutizzazione (crisi emolitica). L'esacerbazione dell'infezione cronica, le infezioni intercorrenti, la vaccinazione, il trauma mentale, il surriscaldamento e l'ipotermia predispongono allo sviluppo di una crisi emolitica. La malattia viene solitamente rilevata in tenera età se una malattia simile è presente nei parenti. Il primo sintomo che dovrebbe allertarvi è l’ittero prolungato nel tempo. Molto spesso, le prime manifestazioni della malattia vengono rilevate negli adolescenti o negli adulti, poiché compaiono fattori più provocatori. Al di fuori del periodo di esacerbazione, i reclami potrebbero essere assenti. Il periodo di esacerbazione è caratterizzato da deterioramento della salute, vertigini, debolezza, affaticamento, palpitazioni e aumento della temperatura corporea. L'ittero (giallo limone) è il principale e può essere l'unico segno della malattia per lungo tempo. L'intensità dell'ittero dipende dalla capacità del fegato di coniugare la bilirubina libera con l'acido glucuronico e dall'intensità dell'emolisi. A differenza dell'ittero meccanico e parenchimale di origine emolitica, non è caratterizzato dalla comparsa di feci scolorite e di urine color birra. La bilirubina non viene rilevata con l’esame delle urine, poiché la bilirubina libera non passa attraverso i reni. Le feci diventano marrone scuro a causa dell'aumento dei livelli di stercobilina. È possibile che la colelitiasi si manifesti sullo sfondo di una tendenza alla formazione di calcoli con lo sviluppo di colecistite acuta. Quando la via biliare comune è ostruita da un calcolo (coledocolitiasi), al quadro clinico si aggiungono segni di ittero ostruttivo ( pelle pruriginosa bilirubinemia, presenza di pigmenti biliari nelle urine, ecc.). Un segno caratteristico della microsferocitosi ereditaria è la splenomegalia. La milza viene palpata 2-3 cm sotto l'arco costale. Con emolisi prolungata, si pronuncia splenomegalia, che si manifesta con pesantezza nell'ipocondrio sinistro. Il fegato, in assenza di complicanze, è solitamente di dimensioni normali; raramente, in alcuni pazienti con decorso prolungato della malattia, può aumentare. Oltre all'ittero e alla splenomegalia, si può notare un'espansione dei confini dell'ottusità cardiaca relativa, del soffio sistolico e dei toni ovattati. All'esame potrebbe esserci patologie ossee(difficoltà nella crescita e nella disposizione dei denti, palato alto, naso a sella, cranio a torre con orbite strette) e segni di ritardo dello sviluppo. I livelli di emoglobina sono generalmente invariati o moderatamente ridotti. Un forte aumento dell'anemia si osserva durante le crisi emolitiche. Nelle persone anziane si possono osservare ulcere trofiche della gamba di difficile guarigione, causate dalla rottura e agglutinazione dei globuli rossi nei capillari periferici dell'arto. Le crisi emolitiche compaiono sullo sfondo di un'emolisi costantemente in corso e sono caratterizzate da un forte aumento delle manifestazioni cliniche. Allo stesso tempo, a causa della massiccia disgregazione dei globuli rossi, compaiono aumenti della temperatura corporea, dispepsia, dolori addominali e aumenta l'intensità dell'ittero. La gravidanza, l'ipotermia e le infezioni intercorrenti provocano lo sviluppo di crisi emolitiche. In alcuni casi, le crisi emolitiche non si sviluppano nel corso della malattia.
QUADRO EMATOLOGICO. Lo striscio di sangue mostra microcitosi e un gran numero di microsferociti. Aumenta anche il numero dei reticolociti. Il numero di leucociti e piastrine rientra nei limiti normali. Durante le crisi emolitiche si osserva leucocitosi neutrofila con spostamento a sinistra. Nel midollo osseo si osserva iperplasia della linea eritroide. La bilirubinemia non è espressa. Il livello della bilirubina indiretta è in media di 50-70 µmol/l. Aumenta il contenuto di stercobilina nelle feci e di urobilina nelle urine.
DIAGNOSI. La diagnosi di microsferocitosi ereditaria viene posta sulla base del quadro clinico e degli esami di laboratorio. È obbligatorio esaminare i parenti per segni di emolisi e microsferocitosi senza manifestazioni cliniche.
DIAGNOSTICA DIFFERENZIALE. Nel periodo neonatale, la malattia di Minkowski-Choffard deve essere differenziata dall'infezione intrauterina, dall'atresia biliare, dall'epatite congenita e dalla malattia emolitica del neonato. Nell'infanzia - con emosiderosi, leucemia, epatite virale. L'eritromielosi acuta viene spesso confusa con una crisi emolitica, accompagnata da anemia, leucocitosi con spostamento a sinistra, splenomegalia, iperplasia della linea eritroide in midollo osseo. La diagnosi differenziale della microsferocitosi ereditaria con anemia emolitica autoimmune prevede l'esecuzione del test di Coombs, che consente la determinazione degli anticorpi fissati sugli eritrociti, caratteristici dell'anemia autoimmune. È necessario distinguere il gruppo delle anemie emolitiche non sferocitiche dalle microsferocitosi ereditarie. Queste malattie sono caratterizzate da deficit enzimatico negli eritrociti, assenza di sferocitosi, resistenza osmotica normale o leggermente aumentata degli eritrociti, aumento dell'autoemolisi e iperglicemia che non può essere corretta. Spesso, per la diagnosi differenziale, viene utilizzata la curva Price-Jones (una curva che riflette la dimensione dei globuli rossi), lungo la quale nella microsferocitosi ereditaria si verifica uno spostamento verso i microsferociti.
TRATTAMENTO. La splenectomia è l’unico metodo di trattamento efficace al 100% per i pazienti con microsferocitosi ereditaria. Nonostante il fatto che persista la diminuzione della resistenza osmotica e della microsferocitosi negli eritrociti, i fenomeni di emolisi vengono interrotti, poiché a seguito della splenectomia viene rimosso il principale trampolino di lancio per la distruzione dei microsferociti e tutte le manifestazioni della malattia scompaiono. Le indicazioni alla splenectomia sono frequenti crisi emolitiche, grave anemia nei pazienti e infarto splenico. Spesso, se un paziente ha colelitiasi, viene eseguita contemporaneamente la colecistectomia. Nei pazienti adulti con flusso lieve malattia e processo di compensazione, le indicazioni alla splenectomia sono relative. La preparazione preoperatoria comprende la trasfusione di globuli rossi, soprattutto nei casi di anemia grave, e la terapia vitaminica. L'uso di farmaci glucocorticoidi nel trattamento della microsferocitosi ereditaria non è efficace.
PREVISIONE. Il decorso della microsferocitosi ereditaria è raramente grave e la prognosi è relativamente favorevole. Molti pazienti vivono fino a tarda età. I coniugi, uno dei quali ha la microsferocitosi ereditaria, dovrebbero essere consapevoli che la probabilità che si verifichi la microsferocitosi nei loro figli è leggermente inferiore al 50%.
Anemia emolitica ereditaria associata a deficit enzimatico (enzimopatie).
Il gruppo delle anemie emolitiche ereditarie non sferocitiche è ereditato in modo recessivo. Sono caratterizzati da una forma normale degli eritrociti, una resistenza osmotica normale o aumentata degli eritrociti e nessun effetto derivante dalla splenectomia. Una carenza di attività enzimatica porta ad una maggiore sensibilità dei globuli rossi agli effetti di farmaci e sostanze di origine vegetale.
Anemia emolitica acuta associata a deficit di glucosio-6-fosfato deidrogenasi (G-6-PDH).
Si verifica più spesso; secondo l'OMS, circa 100 milioni di persone nel mondo hanno una carenza di glucosio-6-fosfato deidrogenasi. Il deficit di G-6-FDG influisce sulla sintesi di ATP, sul metabolismo del glutatione e sullo stato di protezione dei tioli. È più diffuso tra i residenti dei paesi mediterranei dell'Europa (Italia, Grecia), Africa e America Latina.
PATOGENESI. Negli eritrociti con ridotta attività G-6-PD, la formazione di NADP e il legame dell'ossigeno diminuiscono, così come diminuisce la velocità di riduzione della metaemoglobina e diminuisce la resistenza agli effetti di vari potenziali ossidanti. Gli agenti ossidanti, compresi i farmaci, in tale eritrocita riducono il glutatione ridotto, che a sua volta crea le condizioni per la denaturazione ossidativa degli enzimi, dell'emoglobina e dei componenti costitutivi della membrana eritrocitaria e comporta emolisi intravascolare o fagocitosi. È noto che più di 40 farmaci, senza contare vaccini e virus, hanno il potenziale di causare emolisi intravascolare acuta in soggetti con attività G6PD insufficiente. L'emolisi di tali globuli rossi può anche essere causata da intossicazioni endogene e da numerosi prodotti vegetali.
Esempi di farmaci e prodotti che possono potenzialmente causare emolisi: chinino, delagil, streptocide, bactrim, promizol, furatsilina, furazolidone, furagina, isoniazide, cloramfenicolo, aspirina, acido ascorbico, colchicina, levodopa, nevigramon, blu di metilene, prodotti erboristici (fave , piselli, felce maschio, mirtilli, mirtilli).
QUADRO ANATOMICO-PATOLOGICO. Si osservano ittero della pelle e degli organi interni, spleno- ed epatomegalia, moderato gonfiore e ingrossamento dei reni. L'esame microscopico rivela cilindri contenenti emoglobina nei tubuli renali. Nella milza e nel fegato viene rilevata una reazione dei macrofagi con la presenza di emosiderina nei macrofagi.
CLINICA. Il deficit di G6PD si osserva prevalentemente nei maschi che hanno un singolo cromosoma X. Nelle ragazze, le manifestazioni cliniche si osservano principalmente nei casi di omozigosi.
Esistono 5 forme cliniche di deficit di G-6-PD negli eritrociti:
l'emolisi intravascolare acuta è una forma classica di deficit di G-6-PD. Trovato ovunque. Si sviluppa a seguito dell'assunzione di farmaci, vaccinazioni, acidosi diabetica, in connessione con un'infezione virale;
favismo associato al consumo o all'inalazione di polline di alcuni legumi;
malattia emolitica dei neonati, non associata a emoglobinopatia, con incompatibilità di gruppo e Rh;
anemia emolitica cronica ereditaria (non sferocitica);
forma asintomatica.
Una crisi emolitica può essere scatenata da analgesici, alcuni antibiotici, sulfamidici, antimalarici, farmaci antinfiammatori non steroidei, farmaci chemioterapici (PASK, furadonina), prodotti erboristici (baccelli, legumi) e vitamina K, nonché ipotermia e infezioni. Le manifestazioni dell'emolisi dipendono dalla dose degli agenti emolitici e dal grado di deficit di G-6-FDG. 2-3 giorni dopo l'assunzione dei farmaci, si verificano spesso aumento della temperatura corporea, vomito, debolezza, dolore alla schiena e all'addome, palpitazioni, mancanza di respiro e collasso. L'urina diventa di colore scuro (anche nero), a causa dell'emolisi intravascolare e della presenza di emosiderina nelle urine. Un segno caratteristico dell'emolisi intravascolare è l'iperemoglobinemia; in posizione eretta, il siero del sangue diventa marrone a causa della formazione di metaemoglobina. Allo stesso tempo si osserva anche iperbilirubinemia. Aumenta il contenuto di pigmenti biliari nel contenuto duodenale e nelle feci. Nei casi più gravi, i tubuli renali si intasano con i prodotti di degradazione dell'emoglobina, la filtrazione glomerulare diminuisce e si sviluppa un'insufficienza renale acuta. All'esame obiettivo si notano ittero della pelle e delle mucose, splenomegalia e, meno comunemente, ingrossamento del fegato. Dopo 6-7 giorni l'emolisi termina indipendentemente dal fatto che la terapia venga continuata o meno.
QUADRO EMATOLOGICO. Durante i primi 2-3 giorni di crisi emolitica, nel sangue viene rilevata una grave anemia normocromica. Il livello di emoglobina scende a 30 g/l e al di sotto si osservano reticolocitosi e normocitosi. La microscopia dei globuli rossi rivela la presenza di corpi di Heinz (grumi di emoglobina denaturata). Con una crisi pronunciata, si osserva uno spostamento pronunciato della formula dei leucociti a sinistra, fino alle forme giovanili. Nel midollo osseo viene rilevato un germe eritroide iperplastico con sintomi di eritrofagocitosi.
DIAGNOSI. La diagnosi viene posta sulla base del quadro clinico ed ematologico caratteristico dell'emolisi intravascolare acuta, dei dati di laboratorio che rivelano una diminuzione dell'attività enzimatica del G-6-FDG e dell'identificazione di una connessione tra la malattia e l'uso di agenti emolitici .
TRATTAMENTO. Prima di tutto, il farmaco che ha causato l'emolisi dovrebbe essere sospeso. In caso di lieve crisi emolitica vengono prescritti antiossidanti e utilizzati agenti che aiutano ad aumentare il glutatione negli eritrociti (xilitolo, riboflavina). Allo stesso tempo, il fenobarbital viene somministrato per 10 giorni.
Nei casi gravi con segni pronunciati di emolisi, è necessaria la prevenzione dell'insufficienza renale acuta: vengono eseguite terapia infusionale e trasfusione di sangue. Vengono utilizzati farmaci che migliorano il flusso sanguigno renale (aminofillina IV) e diuretici (mannitolo). Nel caso della sindrome DIC viene prescritto il crioplasma eparinizzato. La splenectomia non viene utilizzata per questo tipo di anemia emolitica.
Emoglobinopatie
Le emoglobinopatie sono anomalie ereditarie nella sintesi delle emoglobine umane: si manifestano con un cambiamento nella struttura primaria o con una violazione del rapporto delle normali catene polipeptidiche nella molecola dell'emoglobina. In questo caso, si verifica sempre un danno ai globuli rossi, che si verifica più spesso con la sindrome da anemia emolitica congenita (anemia falciforme, talassemia). Allo stesso tempo, ci sono numerosi casi di trasporto latente di emoglobina anormale. Le emoglobinopatie sono le malattie ereditarie monogeniche più comuni nei bambini. Secondo l’OMS (1983), nel mondo sono circa 240 milioni le persone affette da emoglobinopatie sia strutturali (qualitative) che quantitative (talassemia). Ogni anno nel mondo nascono e muoiono 200mila malati. Prevalenza significativa di emoglobinopatie in Transcaucasia, Asia centrale, Daghestan, Moldavia, Bashkiria. È noto che normalmente l'emoglobina di un adulto è composta da diverse frazioni: emoglobina A, che costituisce la maggior parte, emoglobina F, che costituisce lo 0,1-2%, emoglobina A 2-2,5%.
Talassemia.
Questo è un gruppo eterogeneo di anemie ipocromiche ereditarie di varia gravità, che si basano su una violazione della struttura delle catene globiniche. In alcuni pazienti il principale difetto genetico è il funzionamento anomalo del tRNA nelle cellule, mentre in altri pazienti vi è la delezione del materiale genetico. In tutti i casi, si osserva una diminuzione della sintesi delle catene polipeptidiche dell'emoglobina. Vari tipi di talassemie con diverse caratteristiche cliniche e manifestazioni biochimiche associato a un difetto in qualsiasi catena polipeptidica. A differenza dell'emoglobinopatia, nella talassemia non si verificano disturbi nella struttura chimica dell'emoglobina, ma si verificano distorsioni nei rapporti quantitativi tra emoglobina A ed emoglobina F. I cambiamenti nella struttura dell'emoglobina interferiscono con il normale corso dei processi metabolici nell'eritrocita, quest'ultimo risulta essere funzionalmente difettoso e viene distrutto nelle cellule del sistema reticoloendoteliale. Nella talassemia, il contenuto di HLA negli eritrociti diminuisce. A seconda del grado di riduzione della sintesi di una particolare catena polipeptidica della molecola di emoglobina, si distinguono due tipi principali di talassemia: a e b.
Anemia emolitica- una malattia del sangue abbastanza rara, la cui caratteristica è un accorciamento del ciclo di vita dei globuli rossi (eritrociti). Normalmente un globulo rosso vive in media dai 3 ai 4 mesi, ma in presenza di anemia questo periodo si riduce a due settimane. Ogni giorno una certa parte dei globuli rossi viene sostituita da nuovi globuli prodotti dal midollo osseo rosso.
Se la durata della vita dei globuli rossi viene ridotta, le cellule sostitutive semplicemente non avranno il tempo di maturare. È per questo motivo che la loro concentrazione in sangue perifericoè notevolmente ridotto.
Varietà
Tutti i tipi esistenti di anemia emolitica sono divisi in due grandi gruppi:
- ereditario;
- acquistato.
Forme ereditarie
- Anemia emolitica non sferocitica. In questo caso la causa della distruzione è l'attività difettosa degli enzimi responsabili del loro ciclo vitale;
- Anemia emolitica microsferocitica. La ragione dello sviluppo di questa patologia è la trasmissione di geni mutati, il cui compito principale è sintetizzare le molecole proteiche che formano la parete dei globuli rossi. In caso di progressione dell'anemia emolitica microsferocitica, la struttura dei globuli rossi, la loro attività e la resistenza alla distruzione diminuiscono significativamente;
- cellula falciforme. La ragione della comparsa di una tale malattia ereditaria è una mutazione del gene, il cui compito principale è codificare l'ordine degli aminoacidi nella produzione di emoglobina. Caratteristica malattia - deformazione dell'eritrocito sotto forma di falce. Le cellule colpite non possono più cambiare completamente la loro forma mentre passano attraverso i vasi sanguigni, motivo per cui si verifica una maggiore distruzione;
- talassemia. Questo gruppo di anemie emolitiche si verifica a causa di un'interruzione nel processo di produzione dell'emoglobina.
Moduli acquistati
- Anemia emolitica autoimmune. L'emolisi dei globuli rossi si verifica a causa della formazione di anticorpi che si accumulano sulle loro membrane. Di conseguenza, così rosso cellule del sangue diventano marcati e i macrofagi iniziano a percepirli come agenti estranei. Nell'anemia emolitica acquisita autoimmune, il sistema immunitario umano stesso distrugge i globuli rossi;
- anemia che si verifica a causa del conflitto Rh e della malattia emolitica dei neonati. La ragione della progressione è l'incompatibilità Rh tra il sangue di una donna e il suo feto. Il corpo della futura mamma inizia gradualmente a formare anticorpi contro i globuli rossi del feto, che contengono l'antigene Rh. Di conseguenza, si formano complessi immunitari che portano alla distruzione delle cellule eritrocitarie;
- anemia emolitica traumatica. Ragioni della progressione: vasi sanguigni, presenza di protesi vascolari, cambiamenti nella struttura dei capillari;
- emolisi degli eritrociti, che progredisce sotto l'influenza di fattori endogeni ed esogeni;
- emoglobulinemia parossistica acuta notturna.
Eziologia
Fattori eziologici che provocano la progressione dell'anemia autoimmune, falciforme e di altri tipi di anemia emolitica nei bambini e negli adulti:
- varie varietà;
- Malattie autoimmuni;
- lesioni di varia gravità;
- patologie precedentemente patite di natura infettiva;
- ustioni termiche e chimiche;
- interventi chirurgici;
- malattie infettive ();
- amiloidosi degli organi vitali;
- contatto prolungato con sostanze tossiche;
- uso a lungo termine di alcuni gruppi di farmaci sintetici;
- trasfusione di sangue incompatibile per il fattore Rh;
- Conflitto Rhesus durante la gravidanza;
- batterico e così via.
Sintomi
Le anemie emolitiche (falciformi, autoimmuni, non sferocitiche e altre) sono caratterizzate dai seguenti sintomi:
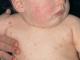
- sindrome da ipertermia. Molto spesso, questo sintomo si manifesta con la progressione dell'anemia emolitica nei bambini. Le temperature aumentano fino a 38 gradi;
- sindrome. La pelle acquisisce tinta gialla, ma allo stesso tempo forte prurito NO. Un sintomo caratteristico è un cambiamento nel colore delle urine. Diventa scuro e ricorda la brodaglia di carne. Gli escrementi non cambiano colore;
- sindrome di ingrossamento del fegato e della milza ();
- sindrome. La pelle e le mucose diventano pallide. Compaiono sintomi di ipossia: mancanza di respiro, debolezza, mal di testa, vertigini, ecc.
Ulteriori sintomi di questa condizione patologica:
- sindrome del dolore nel sito di proiezione del rene;
- dolore addominale;
- dolore al petto;
- cambiamento nelle feci.

Diagnostica
Se avverti sintomi che indicano la progressione di questa malattia, devi visitare istituzione medica per la diagnostica e impostazione precisa diagnosi. Maggior parte metodo informativo rilevamento dell'anemia emolitica è. Con il suo aiuto, il medico sarà in grado di chiarire il numero totale di globuli rossi e la loro qualità. Un esame del sangue può rivelare:
- accelerazione ;
- diminuzione della concentrazione;
- la presenza di globuli rossi deformati;
- diminuzione del numero totale dei globuli rossi;
- aumento della concentrazione.
Tecniche aggiuntive diagnostica:
- puntura del midollo osseo rosso.
Trattamento
Il trattamento dell'anemia emolitica deve essere effettuato solo da uno specialista altamente qualificato. Il punto è questo questa varietà l'anemia è la più difficile da curare, poiché non sempre è possibile eliminare il meccanismo che innesca l'emolisi.
Il piano di trattamento della patologia solitamente prevede le seguenti attività:
- prescrizione di farmaci contenenti vitamina B12 e acido folico;
- trasfusione di globuli rossi lavati. Questo metodo di trattamento viene utilizzato se la concentrazione dei globuli rossi diminuisce a livelli critici;
- trasfusione di plasma e immunoglobuline umane;
- Per eliminare i sintomi spiacevoli e normalizzare le dimensioni del fegato e della milza, si consiglia di utilizzare ormoni glucocorticoidi. Il dosaggio di questi farmaci viene prescritto solo da un medico in base alle condizioni generali del paziente e alla gravità della sua malattia;
- per l'anemia emolitica autoimmune, il piano di trattamento è integrato con citostatici;
- A volte i medici ricorrono a metodi chirurgici per curare la malattia. La procedura più comune è la splenectomia.
Il trattamento della patologia viene effettuato solo in condizioni di degenza in modo che i medici possano monitorare costantemente le condizioni generali del paziente e prevenire la progressione di complicazioni pericolose.
Prevenzione
Tutte le misure preventive per questa malattia sono divise in primarie e secondarie. Le misure prevenzione primaria mirano principalmente a prevenire la progressione dell’anemia emolitica. La prevenzione secondaria comprende misure che aiuteranno a ridurre i sintomi di una patologia già progressiva.
Prevenzione primaria dell’anemia autoimmune:
- evitare infezioni concomitanti;
- non soggiornare in luoghi con alti livelli di sostanze tossiche nell'aria, nonché sali di metalli pesanti;
- trattamento tempestivo delle malattie infettive.
In caso di deficit di G-6-FDG non devono essere utilizzati i seguenti gruppi di farmaci poiché provocano emolisi:
- sulfamidici;
- farmaci antimalarici;
- farmaci antitubercolari;
- antidolorifici;
- antipiretici;
- farmaci antibatterici;
- medicinali di altri gruppi: acido ascorbico, blu di metilene, ecc.
La prevenzione secondaria della malattia consiste nel trattamento tempestivo e completo delle patologie infettive che possono provocare una esacerbazione dell'anemia emolitica. A tal fine, si raccomanda di sottoporsi regolarmente a esami preventivi da parte di specialisti, nonché di sottoporsi a tutti gli esami necessari.
Tutto quello che c'è scritto nell'articolo è corretto dal punto di vista medico?
Rispondi solo se hai comprovate conoscenze mediche
ANEMIA EMOLITICA (anemia emolitica; greco sangue haima + distruzione della lisi, dissoluzione; anemia) è il nome di un gruppo di malattie la cui caratteristica comune è l'aumento della distruzione dei globuli rossi, che causa da un lato anemia e istruzione avanzata prodotti di degradazione degli eritrociti, dall'altro - eritrocitopoiesi potenziata in modo reattivo. Un aumento dei prodotti di degradazione dei globuli rossi si manifesta clinicamente con ittero (vedi) color limone, un aumento dei livelli ematici di bilirubina indiretta (non coniugata) (vedi Iperbilirubinemia) e ferro sierico, pleiocromia della bile e delle feci, urobilinuria ( Vedere). Con l'emolisi intravascolare (vedi), si verificano inoltre iperemoglobinemia (vedi Emoglobinemia), emoglobinuria (vedi), emosiderinuria. L'aumento dell'eritropoiesi è evidenziato dalla reticolocitosi e dalla policromatofilia nel sangue periferico, dall'eritronormoblastosi del midollo osseo.
Classificazione
Divisione di G. a. per acuto e cronico si è rivelato inaccettabile, perché acuto e cronico si possono osservare varianti all'interno delle stesse forme di G. a. Anche l'opposizione a G. non è giustificata. con localizzazione intracellulare e intravascolare dell'emolisi, poiché con la stessa forma di G. a. Può verificarsi emolisi intracellulare e intravascolare. La divisione di G. a. non è priva di convenzione. su quelli causati da fattori emolitici endo- ed esoeritrociti; ad esempio, nell'emoglobinuria parossistica notturna, il difetto principale è localizzato nella membrana eritrocitaria e il fattore emolitico (complemento) è localizzato all'esterno dell'eritrocita.
La divisione più giustificata delle anemie emolitiche in due gruppi principali è l'anemia emolitica ereditaria (congenita) e quella acquisita. Ereditario G. a. può essere causato da una patologia della membrana eritrocitaria o dalla struttura o sintesi dell'emoglobina, nonché da una carenza di uno degli enzimi eritrocitari. Ereditario G. a. sono accomunati su base genetica, ma differiscono significativamente nell'eziologia, nella patogenesi e nel quadro clinico. Per acquisito G. a. includere grande gruppo anemie immunoemolitiche, un gruppo di membranopatie acquisite, ecc.
Si propone quanto segue classificazione di G. a. (Yu. I. Lorie, 1967; L. I. Idelson, 1975):
I. Ereditario (congenito)
1. Membranopatia degli eritrociti:
a) microsferocitico;
b) ovalocitico;
c) acantocitico.
2. Enzimopenico (enzimopenico):
a) associato a una carenza di enzimi del ciclo dei pentoso fosfati;
b) associato a una carenza di enzimi glicolitici;
c) associato ad una carenza di enzimi coinvolti nella formazione, ossidazione e riduzione del glutatione;
d) associato a una carenza di enzimi coinvolti nell'utilizzo dell'ATP;
e) associato ad una carenza di enzimi coinvolti nella sintesi delle porfirine.
3. Emoglobinopatie:
a) emoglobinopatie qualitative;
b) talassemia.
II. Acquistato
1. Anemia immunoemolitica:
a) autoimmune;
b) isoimmune.
2. Membranopatie acquisite:
a) emoglobinuria parossistica notturna;
b) anemia a cellule speronate.
3. Associato a danno meccanico ai globuli rossi:
a) marzo Emoglobinuria;
b) Malattia di Moshkovich (sin. microangiopatica G. a.);
c) che si manifesta durante la sostituzione della valvola cardiaca.
4. Tossico.
ANEMIA EMOLITICA EREDITARIA
Membranopatia dei globuli rossi
Le membranopatie sono associate ad anomalie nei componenti proteici o lipidici della membrana eritrocitaria, che causano cambiamenti nella loro forma e distruzione prematura. In precedenza venivano classificate anche le cosiddette membranopatie G. a. congenite non sferocitiche, che successivamente, a causa dell'identificazione di una carenza di enzimi glicolitici in essi, furono classificate come anemie enzimopeniche.
Anemia emolitica microsferocitica
Anemia emolitica microsferocitica (sin.: microsferocitosi congenita, malattia di Minkowski-Choffard, microcitemia, anemia sferocitica) fu descritta per la prima volta da Vanlair e Masius (S. Vanlair, Masius, 1871) sotto il nome di microcitemia, ma divenne nota come malattia indipendente dopo i lavori classici di O. Minkowski (1900) e Choffard (A. M. Chauffard, 1907).
Statistiche non sviluppato. La malattia è stata descritta in tutte le parti del mondo; È più comune in Europa che in America. A causa dell'uso diffuso della splenectomia nei pazienti con G. microsferocitico, i bordi, di regola, portano a un cuneo, il recupero, è possibile un aumento della frequenza di questa malattia, poiché aumentano le possibilità di trasmetterla per via ereditaria (i pazienti sopravvivono fino all’età fertile).
Eziologia sconosciuto. L'essenza del difetto nella membrana eritrocitaria non è stata stabilita in modo definitivo. La malattia viene ereditata con modalità autosomica dominante.
Patogenesi. Importante importanza nello sviluppo di G. a. conferiscono un ridotto contenuto di proteine actomiosina-simili nella membrana eritrocitaria e una parziale privazione della membrana di fosfolipidi e colesterolo, che provoca una diminuzione della superficie totale degli eritrociti, una diminuzione del suo indice e favorisce la trasformazione degli eritrociti in microsferociti. Un ruolo secondario è attribuito all'aumento della permeabilità della membrana agli ioni sodio, che potenziano l'attività dell'adenosina trifosfatasi e aumentano l'intensità dei processi di glicolisi. In condizioni di lento flusso sanguigno splenico con bassi livelli di pH e glucosio, l’invecchiamento dei globuli rossi accelera. Inoltre, i microsferociti, che hanno perso la plasticità dei normali eritrociti, vengono ritardati puramente meccanicamente quando passano dalla polpa della milza ai sinusoidi venosi davanti ai pori stretti (diametro fino a 3,5 micron) tra le cellule endoteliali che rivestono la loro superficie. Ciò spiega la cessazione dell'aumento dell'emolisi dopo la splenectomia, nonostante la conservazione della forma microsferocitaria da parte degli eritrociti.
Quadro clinico. Nonostante la natura congenita del G. microsferocitico, le sue prime manifestazioni si notano solitamente nei bambini più grandi e adolescenza, sebbene siano stati descritti casi della malattia sia nei neonati che negli anziani. Il decorso della malattia è molto variabile: da subclinico a grave, con frequenti crisi emolitiche. I disturbi possono essere assenti (come dice Shoffar, "i pazienti sono più gialli che malati") o possono essere causati da ipossia anemica, attacchi di coliche biliari. Il sintomo principale è l'ittero della pelle, della sclera e delle mucose di intensità variabile e inconsistente. L'ittero è accompagnato dalla secrezione di feci intensamente colorate e di urine scure. La milza è costantemente ingrossata, il fegato è ingrossato nella metà dei pazienti.
Alcuni pazienti possono presentare anomalie congenite: cranio a torre, palato gotico, bradi o polidattilia, costole cervicali, strabismo, malformazioni del cuore e dei vasi sanguigni e altri (la cosiddetta costituzione emolitica). Con lo sviluppo della malattia nella prima infanzia, sulla radiografia del cranio si nota un'espansione degli spazi diploici. L'anemia è solitamente moderata, talvolta assente a causa della compensazione della lieve emolisi con l'aumento dell'eritropoiesi. Anche con un'emolisi prolungata e intensa, l'eritropoiesi rimane attiva. Le crisi emolitiche gravi si verificano più spesso nelle donne. Si sviluppano gradualmente, nell'arco di 7-10 giorni, e sono solitamente provocati da infezioni e parto. A volte l'anemia è aggravata dall'improvviso sviluppo del cosiddetto. crisi aplastiche (aregenerative), che sono caratterizzate dalla scomparsa dei reticolociti dal sangue e degli eritronormoblasti dal midollo osseo, una forte diminuzione dell'ittero e altri segni di emolisi. In alcuni pazienti, le crisi sono accompagnate da leucemia e trombocitopenia. In genere, le crisi aplastiche durano 7-10 giorni, a volte fino a 2 mesi.
In alcuni pazienti si formano ulcere bilaterali sulla pelle delle gambe nella zona dei condili interni; guariscono solo dopo la splenectomia. Il meccanismo della loro insorgenza non esclude la possibilità di microtrombosi da parte degli sferociti. Un esame del sangue rivela la microsferocitosi (sferocitosi) degli eritrociti: diametro medio gli eritrociti si riducono a 6 micron o meno, il loro spessore aumenta a 2,5-3 micron, il volume medio degli eritrociti e, di conseguenza, il contenuto medio di emoglobina in un eritrocita sono generalmente normali o leggermente aumentati. I microsferociti possono essere rilevati mediante un normale esame di uno striscio di sangue colorato: hanno l'aspetto di cellule piccole e intensamente colorate senza schiarimento centrale. Oggettivamente, i microsferociti vengono determinati mediante eritrocitometria (vedi): la parte superiore della curva di Price-Jones si sposta a sinistra (verso i microciti), la base della curva si allarga a causa dell'anisocitosi. Il contenuto di reticolociti è costantemente aumentato (fino al 20% o più). La puntura del midollo osseo rivela un'iperplasia pronunciata della linea eritroblastica con un aumento del numero di mitosi e segni di maturazione accelerata. La resistenza osmotica dei microsferociti è nettamente ridotta: l'emolisi può iniziare a una concentrazione di cloruro di sodio vicina a quella fisiologica (0,70-0,75%). La resistenza meccanica dei microsferociti è 4-8 volte inferiore a quella degli eritrociti normali. L'eritrogramma acido (vedi) è caratterizzato da un brusco spostamento del massimo principale a destra, un aumento della durata totale dell'emolisi. Dopo aver lavato gli eritrociti dal plasma, il massimo principale dell'eritrogramma acido si sposta a sinistra, la durata dell'emolisi si riduce; ciò consente di ipotizzare la presenza nel plasma di una sostanza che inibisce l'emolisi dei microsferociti. Con forme cancellate di G. microsferocitico a. la stabilità osmotica degli eritrociti va determinata con la loro preliminare incubazione giornaliera a t° 37° (diminuisce in misura molto maggiore rispetto a quella degli eritrociti di individui sani). Allo stesso modo si esamina l'eritrogramma acido degli eritrociti lavati dei pazienti dopo incubazione a t° 37°. Entro 48 ore. L'emolisi spontanea (autoemolisi) è in media del 50%, mentre negli individui sani non supera il 5%. La durata della vita dei microsferociti nel flusso sanguigno è significativamente ridotta. Allo stesso tempo, i tempi di circolazione degli eritrociti normali nel flusso sanguigno dei pazienti, così come i tempi di circolazione dei microsferociti trasfusi a riceventi precedentemente sottoposti a splenectomia, rimangono normali. Ciò conferma l'assenza di fattori di emolisi plasmatica nei microsferociti G. a. Ciò è evidenziato anche dai risultati degli studi sui sieroli: il test di Coombs diretto (vedi reazione di Coombs) è sempre negativo, il test indiretto è positivo nelle pazienti sensibilizzate a seguito di una gravidanza con feto Rh-incompatibile o di trasfusioni di sangue. Il contenuto di emoglobina libera nel plasma è normale. L'emoglobina è sempre di tipo “adulto” (A); solo nei neonati con G. microsferocitica a. rilevare i livelli di emoglobina F adeguati all'età. I livelli di bilirubina sierica sono sempre elevati, principalmente a causa della frazione indiretta (non coniugata). La gravità della bilirubinemia non corrisponde sempre all'intensità dell'emolisi: con una buona funzionalità epatica che secerne bilirubina, può rimanere insignificante. L'escrezione giornaliera di urobilinogeno nelle feci e di urobilina nelle urine aumenta in modo significativo.
Complicazioni. In circa il 30-40% dei pazienti, più spesso con emolisi intensa e prolungata, si formano calcoli pigmentati nelle vie biliari, causando attacchi di coliche biliari. L'ostruzione del dotto biliare comune provoca ittero ostruttivo.
Diagnosi si basa sull'instaurarsi di ittero di tipo emolitico, microsferocitosi e ridotta resistenza osmotica e meccanica degli eritrociti, caratteristico eritrogramma acido. La presenza di microsferocitosi, una ridotta resistenza osmotica degli eritrociti, un test di Coombs diretto negativo e un livello normale di emoglobina libera nel plasma consentono di differenziare i microsferociti G. a. da altri G. a., nonché dall'iperbilirubinemia funzionale.
Trattamento. L'unico metodo che garantisce la cessazione dell'emolisi e il recupero pratico dei pazienti è la splenectomia (vedi). Si ritiene che se il decorso della malattia è calmo, si può astenersi dall'intervento chirurgico. Tuttavia, poiché quasi tutti i pazienti prima o poi sviluppano complicanze, è più corretto operare tutti i pazienti dopo la diagnosi, con la possibile eccezione dei bambini piccoli, degli anziani e dei pazienti con grave patologia cardiovascolare. L'operazione è consentita anche durante la gravidanza (è meglio farlo in combinazione con un taglio cesareo). Le ricadute della malattia dopo la splenectomia si osservano solo in presenza di milze accessorie che non erano state notate durante l'intervento. Tutti i segni di iperemolisi dopo l'intervento chirurgico scompaiono rapidamente, di solito entro 3-4 settimane. La composizione del sangue è completamente normalizzata. La microsferocitosi e la diminuzione della stabilità osmotica dei globuli rossi dopo l'intervento chirurgico persistono per tutta la vita, ma la loro gravità diminuisce leggermente. Le complicazioni e la morte dopo la splenectomia sono rare. Tutte le misure prudenziali per G. a. inefficace. Trasfusione di globuli rossi) deve essere utilizzata a scopo sostitutivo solo in caso di anemia grave (crisi emolitiche e aplastiche). Trasfusioni di sangue ripetute non sono desiderabili a causa del rischio di isosensibilizzazione. Dopo la splenectomia non sono necessarie trasfusioni di sangue.
Previsione dopo splenectomia, favorevole; se l'intervento viene rifiutato, è dubbio per la possibilità di sviluppare queste complicanze. La capacità dei pazienti di lavorare prima del trattamento dipende dalla gravità dell'anemia e dal grado di compensazione dell'ipossia anemica. I pazienti dovrebbero essere informati sulla probabilità di trasmissione della malattia per via ereditaria (ma anche sulla curabilità della malattia). La mortalità è bassa.
Prevenzione non sviluppato. L’unico metodo per prevenire le complicanze è la splenectomia precoce.
Anemia emolitica ovalocitica
Anemia emolitica ovalocitica (sin. ellissocitico G. a.). Per la prima volta, la presenza di globuli rossi di forma ovale nel sangue umano fu descritta da Dresbach (M. Dresbach, 1904). Il sangue di individui sani contiene fino all'8-15% di ovalociti (fiziol, ovalocitosi). Una percentuale maggiore di ovalociti, i cosiddetti. l'ovalocitosi si riscontra nello 0,02-0,05% dei casi e nel 10-12% di essi si osserva ovalociti G. a.
Eziologia sconosciuto. La malattia viene ereditata con modalità autosomica dominante ed è apparentemente trasmessa da due geni, uno dei quali è legato ai geni del sistema Rh. L'espressione genica varia ampiamente.
Patogenesi causata da un difetto della membrana eritrocitaria. L'unico luogo di distruzione dei globuli rossi è la milza, ma il sequestro non è associato ad un'anomalia della loro forma (gli ovalociti dei portatori non rimangono nella milza e hanno un periodo di circolazione normale).
Quadro clinico, complicanze, trattamento, prognosi- come con G. microsferocitico a.
Diagnosi viene posto sulla base della predominanza dei globuli rossi di forma ovale nel sangue periferico, tenendo conto dei sintomi di G. a. Il volume medio degli eritrociti, la concentrazione e il contenuto di emoglobina negli eritrociti sono normali. Nell'aspirato di midollo osseo, le cellule della fila rossa acquisiscono una forma ovale allo stadio di normoblasti policromatofili. La resistenza osmotica degli eritrociti è solitamente normale e diminuisce leggermente durante l'incubazione. Il test di autoemolisi non è potenziato. La durata della vita dei globuli rossi è ridotta, ma quando trasportano ovalociti è normale. Sierolo, reazioni, indicatori del metabolismo dei pigmenti - come nel microsferocitico G. a.
Anemia emolitica acantocitica
L'anemia emolitica acantocitica prende il nome dalla forma degli eritrociti: gli acantociti (greco, spina di akantha, colonna vertebrale) hanno 5-10 escrescenze lunghe e strette a forma di punta sulla superficie. Il contenuto di fosfolipidi e colesterolo nella membrana eritrocitaria è normale, ma si notano cambiamenti nelle frazioni fosfolipidiche: un aumento della sfingomielina e una diminuzione della fosfatidilcolina.
Eziologia. Acantocitico G. a. - malattia rara Presto infanzia associata ad assenza congenita di beta lipoproteine (vedi Abetalipoproteinemia). Ereditato con modalità autosomica recessiva.
Patogenesi. La formazione di acantociti e la loro anormalità fosfolipidica è associata alla presenza di eritrociti nel patol, nel plasma - nei giovani eritrociti i cambiamenti morfologici e biochimici sono minimi. Non ci sono proteine B (la componente proteica delle lipoproteine p), trigliceridi nel plasma; i livelli di colesterolo sono generalmente inferiori a 50 mg/100 ml, i fosfolipidi - inferiori a 100 mg/100 ml.
Quadro clinico caratterizzato da una combinazione di moderato G. a. e steatorrea (vedi) con malassorbimento selettivo dei grassi. Il quadro ematico è caratteristico: i globuli rossi hanno proiezioni lunghe, strette, a forma di punta, la loro aspettativa di vita è ridotta e viene rilevata reticolocitosi. La resistenza osmotica degli eritrociti è normale, l'autoemolisi dopo incubazione a t° 4° e 37° è nettamente aumentata, corretta con l'aggiunta di vitamina E [Brain (M. S. Brain), 1971]. Non ci sono proteine B e trigliceridi nel plasma, il contenuto di colesterolo e fosfolipidi è ridotto.
Complicazioni. Retinite pigmentosa (che porta alla cecità) e neuropatia atassica.
Diagnosi posto sulla base di un caratteristico cuneo, immagine, rilevamento della forma acantocitica degli eritrociti, reticolocitosi, riduzione della durata della vita degli eritrociti.
Trattamento non sviluppato. La somministrazione di vitamina E non è efficace.
Previsione sfavorevole rispetto alla vita.
Anemia enzimatica
G.a. svilupparsi a causa di una carenza di vari enzimi eritrocitari. In base alla carenza di alcuni sistemi enzimatici, si distinguono diversi gruppi di enzimopatie:
G. a.associato a un deficit degli enzimi del ciclo pentoso-fosfato (deficit di glucosio-6-fosfato deidrogenasi, deficit di 6-fosfogluconato deidrogenasi);
G. a. associato a una carenza di enzimi glicolitici (carenza di piruvato chinasi, triosefosfato isomerasi, 2,3-difosfoglicerato mutasi, ecc.);
G. a., associato ad un deficit di enzimi coinvolti nella formazione, ossidazione e riduzione del glutatione (deficit di sintetasi, reduttasi e perossidasi);
G. a., associato a deficit di adenosina trifosfatasi, adenilato chinasi, ribofosfato-pirofosfato chinasi, cioè enzimi coinvolti nell'utilizzo dell'ATP;
G. a., associato ad una carenza di enzimi coinvolti nella sintesi delle porfirine. Si tratta dell'uroporfiria eritropoietica e della protoporfiria eritropoietica (vedere Anemia enzimatica).
Emoglobinopatie
G. a., associato a una violazione della struttura o sintesi dell'emoglobina. Esistono emoglobinopatie causate da un'anomalia nella struttura primaria dell'emoglobina, o qualitative, ad esempio, anemia falciforme (vedi), e causate da una violazione della sintesi delle catene di emoglobina, o talassemia quantitativa (vedi), con alcune di loro emoglobina H, emoglobina Bart e così via.
ANEMIA EMOLITICA ACQUISITA
Anemie immunoemolitiche
L’anemia immunoemolitica è caratterizzata dalla presenza nel sangue di anticorpi contro gli antigeni dei globuli rossi propri o trasfusi (donatore).
L'anemia emolitica autoimmune può essere causata dalla presenza di autoanticorpi caldi, agglutinine fredde, emolisine bifasiche, nonché autoanticorpi che compaiono durante l'assunzione di determinati farmaci.
Anemie autoimmunoemolitiche causate da autoanticorpi caldi
Anemia autoimmunoemolitica causata da autoanticorpi caldi (sin.: p rioacquisito G. a., ittero emolitico acquisito di tipo Guyem-Vidal, anemia immunoemolitica), hanno due forme: idiopatica e sintomatica (che si sviluppa più spesso sullo sfondo di tumori del tessuto linfoide e grandi collagenosi, ad esempio nel lupus eritematoso sistemico).
Questa forma rappresenta ca. Il 25% di tutti i G. a. Sono colpite persone di qualsiasi età; le donne hanno una probabilità leggermente maggiore rispetto agli uomini. Il rapporto tra forme idiopatiche e sintomatiche è 1:1.
Eziologia sconosciuto. La natura acquisita della malattia è evidenziata dall'assenza di casi familiari. In alcuni pazienti, lo sviluppo della malattia è associato all'assunzione di metildopa.
Patogenesi: Esistono due teorie per la formazione di autoanticorpi: 1) un cambiamento primario nella membrana eritrocitaria con la formazione di un nuovo antigene o l'esposizione di un antigene nascosto (profondo) e la successiva reazione del sistema immunologico; 2) cambiamento primario nelle cellule (mutazione somatica) immunol, sistemi con formazione di anticorpi contro i normali antigeni eritrocitari. Gli autoanticorpi caldi, secondo le loro proprietà sierologiche, appartengono molto spesso ad agglutinine incomplete; basato su immunochimici Gli studi (utilizzando sieri antiglobulinici monospecifici) li classificano come immunoglobuline G (IgG), a volte vengono rilevate simultaneamente le immunoglobuline M e A (IgM e IgA). L'emolisi degli eritrociti su cui sono fissati gli anticorpi avviene attraverso la loro frammentazione o eritrofagocitosi. La distruzione dei globuli rossi avviene nella milza, nel midollo osseo, nei linfonodi e nel fegato. In alcuni pazienti, i globuli rossi vengono distrutti direttamente nel flusso sanguigno; In questa parte dei pazienti vengono rilevate anche le emolisine complete.
Quadro clinico: l'esordio della malattia è spesso graduale, ma può essere anche acuto, con quadro di rapida emolisi e coma anemico (il cosiddetto G. A. Lederer acuto). Il decorso è solitamente cronico, con periodi di esacerbazioni. I reclami dei pazienti sono principalmente dovuti all'ipossia anemica. La pelle è pallida, itterica e talvolta l'acrocianosi è chiaramente espressa. L'ittero può essere di varia intensità ed è accompagnato da pleiocromia fecale e urobilinuria. Il contenuto sierico della bilirubina indiretta è aumentato; con l'emolisi in rapida progressione, accompagnata da necrosi epatica anemica, aumenta anche la frazione coniugata della bilirubina. La milza è leggermente o moderatamente ingrandita; nella forma sintomatica è possibile una grave splenomegalia (a causa della malattia di base). Il fegato è ingrossato approssimativamente nei pazienti cronici. Gli esami del sangue rivelano anemia normo o ipercromica, elevata reticolocitosi, talvolta normoblasti, forte anisocitosi degli eritrociti, presenza di microsferociti e macrociti; ci sono frammenti di eritrociti che eritrofagocitosi monociti. Si osserva spesso l'autoagglutinazione dei globuli rossi. Il volume medio degli eritrociti è solitamente aumentato, la loro resistenza osmotica è ridotta e dopo l'incubazione degli eritrociti diminuisce ancora di più (ma meno che con i microsferociti G. a.). Dopo l'incubazione degli eritrociti, viene potenziata anche la loro autoemolisi. L'emoglobina libera nel plasma è spesso elevata, soprattutto in presenza di emolisine nel sangue e al culmine delle crisi emolitiche. Con un'emoglobinemia significativa e prolungata, il livello di aptoglobina plasmatica diminuisce e nelle urine possono comparire emoglobina ed emosiderina. La durata della vita dei globuli rossi, sia i propri che quelli trasfusi da un donatore, è ridotta, spesso in modo significativo. Il numero dei leucociti è normale o ridotto durante i periodi cronici, ma durante le esacerbazioni della malattia si può osservare una significativa leucocitosi neutrofila con uno spostamento a sinistra. Per i sintomi autoimmuni G. a. formula dei leucociti determinata dalla malattia di base. La conta piastrinica è normale o diminuita, a volte bruscamente. Il mielogramma mostra una pronunciata reazione eritronormoblastica. L'eritropoiesi è macronormoblastica, spesso con presenza di megaloblasti, che è associata ad un aumento del consumo di vitamina B12 endogena e di acido folico. Una grave trombocitopenia può portare allo sviluppo di gravi emorragie (sindrome di Fisher-Evans), talvolta contemporaneamente alla leucopenia (pancitopenia immunitaria).
Complicazioni: crisi aplastiche, trombosi che portano ad infarti degli organi interessati; La formazione di calcoli nei dotti biliari è rara.
Diagnosiè diventato possibile dopo l'introduzione del test diagnostico di Coombs. Si basa sulla costituzione dell'acquisita G. a. con localizzazione intracellulare o mista dell'emolisi ed è confermata da un test di Coombs diretto positivo, l'intensità del taglio è diversa, ma può corrispondere alla gravità dell'emolisi. A volte un test diretto è negativo o diventa positivo relativamente tardi nella malattia. La positività al test di Coombs indiretto (rilevazione degli anticorpi liberi nel plasma) non è patognomonica per il G. autoimmune, ma è solitamente causata dalla presenza di isoanticorpi (dopo trasfusioni, durante la gravidanza).
Trattamento: Di solito vengono prescritti ormoni corticosteroidi. La dose iniziale di prednisolone deve essere di almeno 1 mg per 1 kg di peso corporeo al giorno per via orale; nei casi gravi e nell'anemia profonda, la dose viene aumentata a 2-3 dosi per 1 kg di peso, la metà delle quali viene somministrata per via parenterale. Con il miglioramento, la dose del farmaco viene gradualmente ridotta, ma sufficiente a garantire un aumento dell'emoglobina. Dopo la normalizzazione della conta dei globuli rossi, gli ormoni continuano ad essere somministrati a piccole dosi (15-20 mg di prednisolone al giorno); in una condizione di ematolo. in remissione vengono somministrati per altri 2-3 mesi. e solo allora cancellarlo gradualmente. Sali e alcali di potassio vengono prescritti contemporaneamente agli ormoni. Il meccanismo del trattamento azioni degli ormoni nel sistema autoimmune G. a. poco chiaro Si presume un effetto inibitorio sulle cellule immunocompetenti, ma la velocità dell'effetto terapeutico (a volte entro 24-48 ore) indica un effetto diretto sul processo di distruzione del sangue. La terapia ormonale fornisce un cuneo di recupero in circa il 75% dei pazienti. Il test diretto di Coombs rimane positivo per un certo numero di mesi e anni. L'effetto negativo della terapia ormonale può essere spiegato sia dall'incapacità di utilizzare dosi sufficienti di ormoni a causa dello sviluppo di diabete, ipertensione, ecc., sia dalla resistenza ai corticosteroidi. In questi casi è indicata la splenectomia; è efficace in circa la metà dei pazienti operati, ma non esclude recidive tardive di emolisi. Se la terapia corticosteroidea fallisce, si utilizzano anche gli immunosoppressori (6-mercaptopurina, azatioprina, ciclofosfamide, ecc.). Sono stati riportati casi di utilizzo efficace della timectomia nei bambini. Le trasfusioni di sangue (globuli rossi concentrati) sono indicate solo in caso di anemia grave e progressiva. In caso di coma anemico vengono trasfusi fino a 750-1000 ml di sangue contemporaneamente (il donatore viene selezionato utilizzando il test di Coombs indiretto).
Previsione spesso dubbioso, anche se non si può escludere la possibilità di un decorso tranquillo a lungo termine e addirittura di una guarigione spontanea. La capacità di lavorare dei pazienti prima del trattamento è stata persistentemente ridotta. I sintomi prognosticamente sfavorevoli includono la presenza di grave trombocitopenia, un test di Coombs indiretto positivo ed emolisina nel siero. Le cause immediate di morte possono essere emolisi incontrollabile, sanguinamento trombocitopenico e trombosi.
Anemia emolitica causata da autoanticorpi freddi
Esistono forme idiopatiche e sintomatiche. Il sintomo si sviluppa spesso sullo sfondo di alcuni processi linfoproliferativi, mononucleosi infettiva, polmonite da micoplasma (atipica); possibile a qualsiasi età. La forma idiopatica della malattia è rara, più spesso nelle donne e negli anziani.
Eziologia sconosciuto. Non è stato stabilito il meccanismo di formazione delle agglutinine fredde sotto l'influenza di agenti patogeni infettivi.
Patogenesi: gli autoanticorpi freddi vengono fissati insieme al complemento sui globuli rossi piccoli vasi parti distali del corpo (se raffreddate a una temperatura inferiore a 32°).
Un'emolisi evidente si verifica quando il titolo anticorpale è 1: 1000. Le agglutinine fredde hanno specificità sierolica per gli antigeni I o i (quest'ultimo è più comune nella forma sintomatica). Immunohim. dai metodi di ricerca vengono identificati come immunoglobuline M (IgM), meno spesso viene rilevata una combinazione di immunoglobuline M e G (IgM + IgG); le catene X sono responsabili dell'attività emolitica. La distruzione degli eritrociti agglutinati avviene come in letto vascolare e come risultato dell'eritrofagocitosi nella milza, nel fegato e nel midollo osseo (localizzazione mista dell'emolisi). Gli agglutinati nei piccoli vasi compromettono la circolazione sanguigna al loro interno, causando la clinica della sindrome di Raynaud (vedi malattia di Raynaud).
Quadro clinico: il cuneo principale, le manifestazioni della malattia sono generalmente moderatamente espresse G. a. e disturbi derivanti durante il raffreddamento circolazione periferica a seconda del tipo di sindrome di Raynaud. Si osserva acrocianosi e raramente acrogangrena. L'ittero di solito non è intenso. Le dimensioni del fegato e della milza sono normali o leggermente ingrossate. Il decorso della malattia è solitamente cronico e non progressivo. Sono possibili gravi crisi emoglobinuriche. La forma sintomatica si manifesta in modo acuto e termina con una guarigione spontanea. Gli esami del sangue rivelano un'anemia moderata. I globuli rossi sono morfologicamente leggermente modificati, a volte lieve sferocitosi, eritrofagocitosi; una volta raffreddati, i globuli rossi si agglutinano rapidamente; Dopo aver riscaldato il campione di sangue, gli agglutinati dei globuli rossi scompaiono. La resistenza osmotica degli eritrociti è normale o leggermente ridotta. La reticolocitosi è moderata. Il numero di leucociti e piastrine è normale o ridotto. Il ROE può essere notevolmente accelerato. Nel plasma aumentano la bilirubina indiretta e l'emoglobina libera (dopo raffreddamento); l'emoglobina e l'emosiderina possono essere rilevate nelle urine.
Complicazioni può essere causato da un alterato flusso sanguigno nei piccoli vasi (ad esempio, lo sviluppo di cancrena delle dita delle mani e dei piedi).
Diagnosi si basa sull'identificazione di G. a., della sindrome di Raynaud e sulla rilevazione di agglutinine fredde a titolo abbastanza elevato (1: 1.000.000). Il test di Coombs diretto (per il test si preleva il sangue in un contenitore riscaldato a t° 37°) con siero intero antiglobulina è sempre positivo; tra i sieri monospecifici è positivo solo con anti-C.
Trattamento: I corticosteroidi e la splenectomia sono inefficaci. È stato descritto un effetto benefico del leukeran. In caso di anemia profonda sono indicate le trasfusioni di globuli rossi lavati dal plasma (per eliminare il complemento).
Previsione dubbioso riguardo al recupero. La capacità lavorativa può essere mantenuta.
Anemia emolitica causata da emolisine bifasiche
L'anemia emolitica causata da emolisine bifasiche (emoglobinuria parossistica fredda) è una malattia rara che rappresenta il 4,6% di tutte le anemie immunoemolitiche.
Eziologia. La malattia si sviluppa durante le infezioni virali acute, meno spesso durante. sifilide
Patogenesi. L'Emoglobinuria parossistica da freddo si verifica quando nel sangue sono presenti emolisine bifasiche di Donath-Landsteiner che, quando il corpo si raffredda, si depositano sui globuli rossi ed effettuano l'emolisi ad una temperatura di 37°. Le emolisine bifasiche hanno mobilità elettroforetica corrispondente alla frazione gamma; appartengono all'immunoglobulina G (IgG).
Quadro clinico caratterizzato da sintomi di gravi condizioni generali, mancanza di respiro, febbre, mal di testa, dolori muscolari e articolari, nonché segni di rapida emolisi intravascolare (comparsa di urina nera, ittero, anemia). Si osservano spesso vomito incontrollabile di bile e feci molli. La milza e il fegato sono moderatamente ingrossati e sensibili. Una forma lieve di emoglobinuria parossistica da freddo si manifesta con febbricola ed emoglobinuria a breve termine. Gli esami del sangue rivelano una grave anemia normocromica, punteggiatura basofila degli eritrociti, policromasia degli eritrociti, normoblasti, un aumento del numero di reticolociti e leucocitosi neutrofila con spostamento a sinistra, talvolta verso i promielociti e persino i mieloblasti. Viene rilevata iperbilirubinemia (dovuta alla bilirubina non coniugata), un aumento dell'emoglobina a 30-40 mg/100 ml. Il siero del sangue è di colore rosa; in posizione eretta diventa marrone a causa della formazione di metaemoglobina. Nella puntura del midollo osseo c'è un quadro di eritropoiesi reattiva, eritrofagocitosi. Altri studi rivelano emoglobinuria (vedi), pleiocromia della bile, aumento della secrezione di stercobilina nelle feci.
Complicazioni: insufficienza renale, anuria.
Trattamento: attuare misure anti-shock ( farmaci cardiovascolari, morfina, adrenalina, cortina, ossigeno), trasfusione di sangue di un singolo gruppo (250-500 ml), poliglucina (500-1000 ml), alcali sono prescritti per via orale e endovenosa (soluzione al 5% preparata al momento di flebo di bicarbonato di sodio in una quantità totale dose di 500-1000 ml). Per pulire rapidamente il plasma dall'emoglobina, vengono somministrati diuretici osmotici: soluzione al 30% di una preparazione di urea liofilizzata sterile appena preparata in soluzione di glucosio al 10% in una dose totale di 200-300 ml. Sono indicati i glucocorticoidi.
Previsione determinato dall'entità dell'emolisi, dallo stato della funzionalità renale, dalla tempestività e dall'efficacia del trattamento. Se l'esito è favorevole, entro 2-4 settimane. Arriva un cuneo completo, la ripresa. La prognosi è sfavorevole nei casi complicati da anuria e insufficienza renale. Nella forma fulminante è possibile la morte per shock e anossia acuta entro i primi due giorni.
Anemia immunoemolitica indotta da farmaci
L'anemia immunoemolitica indotta da farmaci si verifica quando si verificano reazioni emolitiche indotte da farmaci con la partecipazione di anticorpi.
Eziologia e patogenesi. Durante l'assunzione di alcuni possono comparire autoanticorpi medicinali(penicillina, streptomicina, PAS, indometacina, piramidone, fenacetina, chinino, chinidina, ecc.). Il meccanismo di partecipazione dei farmaci allo sviluppo di G. a. potrebbe essere diverso. Con il meccanismo di sviluppo dell'apteno di G. a. il farmaco si combina con un componente della superficie dei globuli rossi e provoca la formazione di anticorpi antifarmaco di tipo IgG; quando il farmaco viene ripreso, gli anticorpi si fissano sui globuli rossi da esso bloccati. Questo è il meccanismo d'azione della penicillina; in questo caso è possibile che non si osservi la solita reazione allergica alla penicillina. Quando si formano complessi immunitari, il farmaco si lega alla proteina trasportatrice e stimola la formazione di anticorpi di tipo IgM. Il complesso farmaco-anticorpo danneggia la membrana dei globuli rossi, favorisce la fissazione del complemento su di essi, causando emolisi. Questo è il meccanismo d'azione del chinino e della chinidina. Ma il farmaco può indurre la formazione di veri e propri autoanticorpi, come nel caso del calore autoimmune G. a. Questo è il meccanismo d'azione dell'alfa-metildopa (dopegit). Fattore causale Potrebbero esserci anche mebedrolo (mefenammina), clordiazossido (elenio). Dopo la sospensione del farmaco, tutti gli anticorpi scompaiono rapidamente.
Quadro clinico determinato dalla gravità e dalla localizzazione dell'emolisi. Le forme predominanti sono lievi e gravità moderata. La malattia è acuta, con localizzazione mista dell'emolisi. Nel siero si trovano anticorpi che agglutinano i globuli rossi del paziente e degli individui sani (in presenza di questo farmaco).
Diagnosi si basa su dati anamnestici, test di Coombs diretto positivo con sieri monospecifici.
Trattamento si riduce principalmente all'abolizione del farmaco che ha causato G. a. I corticosteroidi sono efficaci solo per l'ipertensione causata dall'alfa-metildopa, ma devono essere usati con cautela a causa del rischio di aumento della pressione sanguigna. Per l'anemia grave sono indicate le trasfusioni di sangue.
Anemie emolitiche isoimmuni può svilupparsi nei neonati con incompatibilità dei sistemi ABO e Rh del feto e della madre (vedi malattia emolitica dei neonati), nonché una complicazione delle trasfusioni di sangue, anche incompatibili con i sistemi ABO, Rh e le sue rare varietà. Si tratta di anemie emolitiche post-trasfusionali (vedi Trasfusione di sangue). Con isoimmune G. a. Gli anticorpi vengono rilevati nel siero durante l'esecuzione del test di Coombs indiretto.
Membranopatie acquisite
Emoglobinuria parossistica notturna
Emoglobinuria parossistica notturna (sin.: Malattia di Strübing-Marchiafava, malattia di Marchiafava-Miceli) è considerata eritrocitopatia acquisita (non sono state identificate forme ereditarie familiari della malattia); si verifica a causa di una mutazione somatica che porta alla comparsa di una popolazione anormale di globuli rossi. L'origine monoclonale di questa popolazione di eritrociti è stata dimostrata. L'emolisi degli eritrociti è causata solo dal complemento, ma è provocata da una varietà di fattori, tra cui la fisiologia (stato di sonno, nelle donne - mestruazioni); la comparsa di emoglobinuria è associata a spostamenti dell'equilibrio acido-base verso l'acidosi nelle condizioni sopra indicate. Gli agenti provocatori possono essere anche infezioni intercorrenti, stati di ipercoagulazione del sangue, farmaci e trasfusioni sia di sangue intero (soprattutto fresco) che plasmatico. L'emoglobinuria parossistica notturna può esordire con il quadro di uno stato emopoiesi ipoplastico; in alcuni casi, il quadro caratteristico della malattia si sviluppa 10-12 o più anni dopo la scoperta dell'ipoplasia emopoietica, talvolta dopo la splenectomia.
Quadro clinico. La malattia è diversa corso lungo. Sullo sfondo dell'emoglobinemia e dell'emosiderinuria si verificano parossismi di emoglobinuria, spesso notturni.
Un esame del sangue rivela una grave anemia ipocromica, una moderata diminuzione del numero di granulociti e piastrine. A causa dell’emosiderinuria prolungata (“diabete ferrico”), i depositi di ferro nel corpo si esauriscono e si sviluppa iposideremia. Si notano sintomi di ittero emolitico: iperbilirubinemia (dovuta alla frazione non coniugata), urobilinuria, pleiocromia biliare, reticolocitosi. Il fegato e la milza spesso non sono ingranditi. Il midollo osseo è iperplastico a causa degli elementi dell'eritropoiesi.
Complicazioni. Durante il periodo delle crisi emoglobinuriche, la malattia è spesso complicata dalla sindrome da ipercoagulazione con successiva trombosi vascolare nel sistema venoso portale, nei vasi addominali, cerebrali, coronarici, nelle donne, inoltre, nei vasi pelvici, che è accompagnata. dolore nell'area della trombosi. La tendenza alla trombosi vascolare è associata all'ingresso nel sangue di sostanze tromboplastiche provenienti da globuli rossi disintegrati. La trombosi è talvolta complicata da attacchi di cuore vari organi; in particolare, la trombosi nel sistema della vena porta porta ad infarti splenici con sviluppo di splenomegalia tromboflebitica e ipertensione portale. In rari casi, si verifica una transizione dall'emoglobinuria parossistica notturna alla sindrome iperplastica e mieloproliferativa - eritromielosi o leucemia mieloblastica acuta.
Diagnosi si effettua mediante specifici esami di laboratorio (test dell'acido e del saccarosio), nonché sulla base di un cuneo, un quadro di continua emolisi intravascolare, spesso accompagnata da parossismi di emoglobinuria notturna. Il test dell'acido, o test di Ham, si basa sulla determinazione della sensibilità degli eritrociti al complemento di siero umano fresco, acidificato mediante l'aggiunta di una soluzione di acido cloridrico allo 0,2% a pH 6,5. Il test è considerato positivo se più del 5% (a volte fino al 50-80%) dei globuli rossi sono emolizzati. Il test del saccarosio, o test di Hartmann-Jenkins, si basa sul fatto che gli eritrociti dei pazienti vengono lisati in una soluzione debole di saccarosio in presenza di complemento. Il campione è considerato positivo quando più del 4% dei globuli rossi analizzati risulta lisato.
Trattamento si tratta di mantenere la conta eritrocitaria a un livello ottimale attraverso trasfusioni sistematiche di globuli rossi lavati 3-5 volte (fiziol, soluzione) o di globuli rossi vecchi di 7-10 giorni (periodo di inattivazione del complemento). La trasfusione di sangue intero fresco e plasma è controindicata perché aumenta l'emolisi. Per l'iposideremia sono indicati gli integratori di ferro in piccole dosi tollerabili individualmente, in combinazione con ormoni anabolizzanti (Nerobol, Retabolil). Per le complicanze trombotiche viene prescritta l'eparina, talvolta in combinazione con la fibrinolisina. I glucocorticoidi (prednisolone) sono controindicati nella fase avanzata della malattia. Nella fase ipoplastica della malattia, è indicato l'intero arsenale di farmaci utilizzati per l'anemia ipoplastica: sono accettabili glucocorticoidi, androgeni, trasfusioni di sangue e trasfusioni di globuli rossi freschi non lavati. In caso di sanguinamento trombocitopenico persistente è indicata la splenectomia.
Previsione serio. La morte può verificarsi nella fase iniziale a causa di coma anemico sullo sfondo di sanguinamento trombocitopenico, nella fase emolitica avanzata - a seguito di complicanze vascolari trombotiche o settiche, in rari casi di leucemia acuta.
Anemia emolitica a cellule speroni
L'anemia emolitica a cellule sperone è stata descritta da J. A. Smith et al. (1964) in pazienti con forme gravi di cirrosi epatica.
Eziologia sconosciuta.
La patogenesi della malattia è associata all'eccesso di colesterolo e all'insufficienza di fosfolipidi nella membrana degli eritrociti.
Quadro clinico, trattamento e prognosi come per i microsferociti G. a.
La diagnosi si basa sul rilevamento dei globuli rossi con numerosi piccoli processi nel sangue.
Anemia emolitica causata da un danno meccanico ai globuli rossi
Emoglobinuria parossistica in marcia
In marcia emoglobinuria parossistica fu descritta per la prima volta da Fleischer (I. Fleischer, 1881), che la osservò in un soldato sano che aveva compiuto un lungo cammino a piedi.
Eziologia e patogenesi. L'emolisi degli eritrociti si sviluppa in giovani fisicamente forti a causa dell'aumento del carico sui muscoli degli arti inferiori durante lunghe camminate, marce, corse, sci, nonché sui muscoli delle braccia durante le tecniche di karate. Secondo Davidson (R. J. L. Davidson, 1964), l'emoglobinuria in marcia si verifica quando si corre su una superficie dura (dopo aver corso su una superficie morbida o aver indossato scarpe con solette elastiche, l'emoglobinuria non si sviluppa negli stessi individui). Un fattore predisponente è l’ipoaptoglobinemia. L'emolisi meccanica si sviluppa localmente nei vasi delle parti del corpo che subiscono una collisione prolungata con una superficie dura (piedi, mani).
Quadro clinico caratterizzato da un decorso lieve della malattia, assenza di febbre ed è dovuto all'intensità dell'emolisi intravascolare. Sono possibili gravi crisi emoglobinuriche; sono più comuni emoglobinemia ed emoglobinuria moderate e una diminuzione dell'aptoglobina sierica. La condizione iniziale dei pazienti è normale. Morphol, non si notano anomalie eritrocitarie.
Diagnosi differenziale con altra emoglobinuria si basa sui dati dell'anamnesi (la connessione della malattia con un fattore meccanico e non con il raffreddamento o l'assunzione di farmaci) e sui risultati dei test degli eritrociti (saccarosio e acido). Si distingue dalla mioglobinuria in marcia (vedi) per l'assenza dolore muscolare, rilevamento dell'emoglobina nelle urine.
Trattamento solitamente non richiesto.
Prevenzioneè cambiare le condizioni fisiche. carichi: a volte è sufficiente cambiare scarpe con scarpe più elastiche e cambiare tecnica di corsa per eliminare completamente l'emolisi dei globuli rossi.
Previsione favorevole.
Malattia di Moshkovich
Malattia di Moshkovich (sin. microangiopatico G. a.) è un concetto di gruppo che denota G. a. in alcune patologie, condizioni causate da danni ai piccoli vasi (arteriole) in combinazione con coagulazione intravascolare disseminata (vedi malattia di Moshkovich).
Anemie emolitiche che si verificano durante la sostituzione della valvola cardiaca
Quando si sostituiscono le valvole cardiache, è possibile sviluppare G. a., che è dovuto a trauma meccanico e rottura della membrana (frammentazione) degli eritrociti inizialmente pieni del paziente. Più spesso si sviluppa con insufficienza valvole artificiali cuore sinistro a causa del passaggio forzato del sangue durante la sistole ventricolare attraverso gli spazi tra la protesi e l'anello della valvola.
Quadro clinico manifestato dall'intensità dell'emolisi intravascolare, che è più pronunciata in comportamento attivo paziente rispetto a quando si comporta in modo severo riposo a letto. Gli esami del sangue rivelano anemia, talvolta ipocromia degli eritrociti, reticolocitosi, emoglobinemia e diminuzione o assenza di aptoglobina nel plasma. Caratteristica è la presenza di morfolo. segni di frammentazione degli eritrociti (schistociti, eritrociti triangolari e a forma di elmo). Sono stati descritti casi con test di Coombs diretto positivo. L'emoglobina e l'emosiderina vengono rilevate nelle urine.
Diagnosi sulla base dell'anamnesi, degli esami del sangue (segni di emolisi intravascolare e frammentazione degli eritrociti) e delle urine (presenza di emoglobina ed emosiderina).
Trattamento. In caso di anemia grave persistente è indicato l'intervento chirurgico con ricostruzione della protesi. Nei casi lievi, si limitano a trasfusioni di sangue ripetute e alla prescrizione di integratori di ferro. I corticosteroidi non sono efficaci.
Anemia emolitica tossica
Eziologia. L'emolisi dei globuli rossi può essere causata da numerose sostanze chimiche. e natura batterica. Da chimica. sostanze, l'emolisi è spesso causata dall'idrogeno dell'arsenico (attraverso l'interazione dei composti dell'arsenico con i gruppi sulfidrilici), piombo, sali di rame (a causa dell'inibizione della piruvato chinasi e di altri enzimi eritrociti), clorati di potassio e sodio, meno comunemente resorcinolo, nitrobenzene, anilina. Sono stati descritti casi di G. a. durante l'ossigenazione iperbarica, dopo punture di api e ragni.
Patogenesi. Il meccanismo dell'emolisi può essere diverso. L'emolisi può verificarsi a causa di un forte effetto ossidativo (come nell'anemia enzimatica), che supera la normalità meccanismi di difesa eritrociti, a causa della ridotta sintesi di porfirine, della comparsa di fattori autoimmuni, ecc. La distruzione degli eritrociti avviene più spesso per via intravascolare. Tossico G.a. possono svilupparsi in malattie infettive. Il meccanismo dell'emolisi in alcuni di essi è noto. Pertanto, Bartonella bacilliformis - plasmodia della malaria penetra nei globuli rossi, che vengono poi eliminati dalla milza. Clostridium welchii produce alfa-tossina-lecitinasi, che interagisce con i lipidi della membrana eritrocitaria per formare lisolecitina emoliticamente attiva. Nella leishmaniosi, l'emolisi è associata a splenomegalia. Sono possibili anche altri meccanismi di emolisi: adsorbimento di polisaccaridi batterici sugli eritrociti con successiva formazione di autoanticorpi, distruzione da parte dei batteri dello strato superficiale della membrana eritrocitaria con esposizione dell'antigene T e poliagglutinabilità degli eritrociti.
Quadro clinico e complicanze. Tossico a valle G. a. può essere acuto e cronico. Nella tossicità acuta G. a. Si verifica emolisi intravascolare, che si manifesta con emoglobinemia, emoglobinuria e nei casi più gravi può essere accompagnata da sintomi di collasso e anuria. Con G. cronico e tossico a. Predomina l'emolisi intracellulare, che porta a epato- e splenomegalia, che è particolarmente pronunciata nella malaria e nella leishmaniosi viscerale.
Trattamento consiste nell'interruzione del contatto con l'agente tossico e nell'uso di antidoti appropriati e, in caso di malattie infettive accompagnate da G. a., nella terapia del processo principale. Nell'anemia grave sono indicate le trasfusioni sostitutive. Con l'anuria la diuresi va mantenuta introducendo liquidi nell'organismo, in particolare soluzioni alcaline. La quantità di liquidi somministrati non deve superare la diuresi giornaliera.
Previsione. Nel decorso acuto della G.a tossica. Possibile morte; Con l'identificazione tempestiva e l'eliminazione della causa dell'emolisi, si osserva il completo recupero. Quando è cronico, il decorso del G. tossico a. dipende anche la prognosi rilevamento precoce cause della malattia e la sua eliminazione. L’emolisi, che accompagna alcune malattie infettive, regredisce quando l’infezione viene trattata.
Dati riassuntivi sulle caratteristiche diagnostiche differenziali di G. a. sono presentati nella tabella.
ANATOMIA PATOLOGICA
Con G.a. a causa della maggiore distruzione dei globuli rossi, si osservano anemia, ittero (vedi), iperplasia del midollo osseo (vedi), ingrossamento della milza (vedi) e del fegato (vedi), emosiderosi (vedi) di organi e tessuti, emorragie multiple e trombosi vascolare , focolai di emopoiesi extramidollare (vedi). Questi cambiamenti sono espressi in misura diversa a seconda della forma di G. a. Per tutte le forme di G. a. rilevare la degenerazione grassa del miocardio, del fegato, spesso necrobiosi e necrosi delle cellule epatiche dipartimenti centrali lobuli, sono possibili cambiamenti cirrotici. Nei piccoli vasi e nei capillari vengono rilevati accumuli di globuli rossi aggregati, talvolta emolizzati.
Si osservano spesso emorragie negli organi e nei tessuti, coaguli di sangue freschi e vecchi nei vasi del sistema portale, nei polmoni, nel cervello, ecc.
Con ereditario G. a. L'autopsia rivela un ittero generale, talvolta una deformazione delle ossa del cranio e spesso ulcere trofiche sulle gambe. Midollo osseo piatto e ossa tubolari succoso, rosso, spesso con una sfumatura arrugginita.
Riso. 2. Milza con anemia emolitica microsferocitica ereditaria. Congestione, riduzione dei follicoli (indicata dalle frecce).

Riso. 3. Midollo osseo delle ossa piatte nell'anemia emolitica ereditaria. Grave iperplasia delle forme nucleari della serie rossa; X600.

Riso. 4. Area di emopoiesi extramidollare nel tessuto adiposo adiacente della ghiandola surrenale nell'anemia emolitica ereditaria, le frecce indicano il tessuto surrenale invariato; X200.

Riso. 5. Milza nell'anemia emolitica autoimmune. Accumulo focale di cellule reticolari (indicate dalle frecce) nella polpa rossa, campi di eritrociti emolizzati della milza; X600.
La milza è notevolmente ingrandita (fino a 3,5 kg), la capsula è ispessita, ci sono aderenze fibrose con i tessuti circostanti, la superficie del taglio è rosso-brunastra, infarti frequenti, escrescenze focali tessuto connettivo con deposizione di prodotti di degradazione dell'emoglobina (i cosiddetti noduli scleropigmentali). Potrebbe esserci un ingrossamento del fegato, dei linfonodi, segni di emopoiesi extramidollare negli organi e nei tessuti sotto forma di noduli rosso scuro (colore Fig. 4). C'è una descrizione di massicce crescite extramidollare di tessuto ematopoietico nel tessuto lungo la colonna vertebrale toracica, che sono esternamente simili alle formazioni tumorali. La cistifellea e i dotti contengono bile densa e scura, spesso calcoli pigmentati. Quando gistol, lo studio del midollo osseo rivela la sua pletora, tra le cellule predominano le cellule della fila rossa: eritroblasti e normoblasti, il numero di mielociti è spesso aumentato (tsvetn. Fig. 3). Si verifica un riassorbimento del tessuto osseo con distruzione focale dello strato osseo corticale. Nella milza, nel fegato, nel midollo osseo, nei linfonodi si osserva costantemente eritrofagia, ma meno pronunciata rispetto alla G. autoimmune acquisita. a. Negli organi e nei tessuti si riscontrano fenomeni di emosiderosi, spesso contemporaneamente ai prodotti privi di ferro della degradazione dell'emoglobina. Gistol, immagine con G. a. microsferocitico: i follicoli della milza sono ridotti, la polpa rossa è nettamente congestionata (colore Fig. 2), i seni venosi nelle aree di congestione sembrano fessure strette. Ci sono molti globuli rossi emolizzati e disintegrati. L'endotelio dei seni è sempre nettamente iperplastico. Nella polpa rossa sono presenti accumuli di globuli rossi immaturi, leucociti segmentati e linfociti. I cambiamenti sclerotici sono espressi a vari livelli.
Nella malattia autoimmune G. a. la milza è generalmente ingrossata, ma meno che nella G. a. ereditaria: il suo peso raramente supera 1 kg. A esame microscopico rivelano riduzione dei follicoli, pletora della polpa, iperplasia dell'endotelio del seno, segni di emolisi degli eritrociti ed eritrofagia pronunciata. Una caratteristica distintiva dell'autoimmune G. a. è la presenza nella milza di significativa iperplasia focale (tsvetn. Fig. 5) o diffusa delle cellule reticolari con comparsa di forme giganti [Rappaport, Crosby (H. Rappaport, W. H. Crosby), 1957; L.A. Danilova, 1960; Dacey (JV Dacie), 1962; Pirofsky (B. Pirofsky), 1969]. Queste cellule presentano un'elevata attività enzimatica, la loro significativa proliferazione corrisponde all'intensità del processo immunologico nei pazienti. Si osserva spesso emosiderosi degli organi. I globuli rossi e i cilindri di emoglobina sono talvolta visibili nei lumi dei tubuli renali. L'iperplasia dei normoblasti e degli eritroblasti si trova nel midollo osseo; ci sono spesso cambiamenti distrofici cellule, si possono sviluppare aree di ipoplasia. Nel midollo osseo sono state descritte formazioni limitate di linfociti maturi [Andre, Duhamel (R. Andre, G. Duhamel), ecc., 1968]. Nella forma sintomatica di G. a. autoimmune, che si sviluppa nella leucemia, si riscontrano anche il morfolo sopra descritto e segni di aumento della distruzione del sangue (A.K. Ageev, 1964).
Con l'emoglobinuria parossistica notturna, all'autopsia si notano segni di anemia, spesso ittero, emorragie multiple di piccoli punti nella pelle, nelle membrane sierose e mucose. Caratterizzato da un aumento delle dimensioni e del peso dei reni, espansione dello strato corticale, che ha un colore bruno-rosso. Trombosi diffuse si riscontrano spesso nel sistema della vena porta, nel cervello e nelle sue membrane. Di conseguenza, in alcuni casi vengono rilevati focolai di rammollimento della sostanza cerebrale, infarti in vari organi e necrosi della parete dell'intestino tenue. A differenza di G. a. con emolisi prevalentemente intracellulare, non vi è alcun ingrossamento pronunciato della milza. Quest'ultimo si nota solo con lo sviluppo di complicanze (trombosi della vena splenica e dei suoi rami intraorganici, attacchi di cuore). Il fegato è leggermente ingrossato. Il midollo osseo delle ossa piatte e tubolari è succoso, di colore rosso scuro e può contenere aree secche di colore rosa chiaro o giallastre. Quando gistol, la ricerca nei reni rivela costantemente massicci depositi di emosiderina nell'epitelio dei tubuli, più spesso nelle loro parti prossimali. Nei lumi dei tubuli possono formarsi accumuli di emoglobina libera e di globuli rossi emolizzati. Vengono descritti i cambiamenti distrofici nell'epitelio e la fibrosi dello stroma renale. La deposizione di emosiderina in altri organi interni si osserva solo quando prescritta ai pazienti grandi quantità trasfusioni di sangue. C'è una degenerazione grassa nel fegato, spesso necrosi nelle parti centrali dei lobuli, soprattutto con trombosi delle vene intraepatiche. Nel midollo osseo, insieme all'iperplasia dei globuli rossi, si possono trovare aree di devastazione di varie dimensioni, rappresentate da stroma edematoso e cellule adipose. Caratterizzato dalla presenza di campi emorragici, espansione del lume dei seni, accumulo di eritrociti emolizzati in essi ed eritrofagia. È possibile aumentare il numero di plasmatici e mastociti. Il numero di granulociti nel midollo osseo è ridotto. Forme degenerative si osservano spesso tra i megacariociti. Il morfolo e i cambiamenti che accompagnano i disturbi circolatori si riscontrano anche in altri organi e tessuti. Nelle vene di varie dimensioni, oltre ai coaguli freschi, si riscontrano anche coaguli organizzati con fenomeni di vascolarizzazione.
Tavolo. Caratteristiche diagnostiche differenziali delle anemie emolitiche
|
Forme di anemia emolitica |
Il meccanismo principale per lo sviluppo dell'emolisi |
Quadro clinico |
Complicazioni |
Dati di laboratorio |
|
|
ANEMIA EMOLITICA EREDITARIA (CONGENATA). |
|||||
|
Membranopatia dei globuli rossi |
|||||
|
Anemia emolitica microsferocitica |
L'assenza di fosfolipidi e colesterolo nella membrana degli eritrociti. La durata della vita dei globuli rossi è ridotta. |
Anemia moderata. Microsferocitosi degli eritrociti, la loro resistenza osmotica è nettamente ridotta. Il test diretto di Coombs è negativo, l'emosiderina è assente nelle urine. Il contenuto di emoglobina libera nel plasma è normale |
Splenectomia |
||
|
Anemia emolitica ovalocitica |
Patologia della membrana eritrocitaria di natura sconosciuta. I globuli rossi vengono emolizzati nella milza |
Ittero, il fegato è costantemente ingrandito, la milza è raramente ingrandita |
Crisi emolitiche e aplastiche; calcoli biliari pigmentati |
Ovalocitosi della maggior parte dei globuli rossi. Gli altri risultati di laboratorio sono gli stessi dell'anemia emolitica acantocitica |
Splenectomia |
|
Anemia emolitica acantocitica |
Nella membrana dei globuli rossi contenuto aumentato sfingomielina e livelli ridotti di fosfatidilcolina. Assenza di betalipoproteine nel plasma. I globuli rossi sono emolizzati nei vasi sanguigni, nel fegato, nella milza |
Ittero, la milza è costantemente ingrandita, il fegato è raramente ingrandito |
Retinite pigmentosa, neuropatia atassica, steatorrea |
Acantocitosi degli eritrociti: il disco eritrocitario ha 5-10 proiezioni lunghe e strette. La resistenza osmotica degli eritrociti non è compromessa; il contenuto di emoglobina libera nel plasma è normale, l'emosiderina è assente nelle urine, il test diretto di Coombs è negativo |
Non sviluppato |
|
Enzimopenico (enzimopenico) |
|||||
|
Anemia emolitica associata a deficit degli enzimi del ciclo del pentoso fosfato |
Carenza di glucosio-6-fosfato deidrogenasi, 6-fosfato gluconato deidrogenasi. |
Crisi emolitiche e aplastiche; calcoli biliari pigmentati |
Agenti disintossicanti, trasfusioni di sangue. |
||
|
Anemia emolitica associata a deficit di enzimi glicolitici |
Carenza di piruvato chinasi, triosofosfato isomerasi, 2,3-difosfoglicerato mutasi, ecc. I globuli rossi vengono emolizzati nella milza |
L'ittero si esprime al momento della crisi emolitica. L'anemia è più pronunciata quando forma acuta che durante hron, flusso. La milza e il fegato talvolta sono ingrossati |
Crisi emolitiche e aplastiche; calcoli biliari pigmentati |
A volte viene rilevata macrocitosi; la resistenza osmotica degli eritrociti è ridotta, durante le crisi il contenuto di emoglobina libera nel plasma aumenta, l'emosiderina può comparire nelle urine |
|
|
Anemia emolitica associata a deficit degli enzimi del ciclo del glutatione |
Carenza di sintetasi, reduttasi, perossidasi. La durata della vita dei globuli rossi è ridotta. I globuli rossi vengono emolizzati nella milza |
L'ittero si esprime al momento della crisi emolitica. L’anemia è più pronunciata nella forma acuta che in quella cronica. La milza e il fegato talvolta sono ingrossati |
Crisi emolitiche e aplastiche; calcoli biliari pigmentati |
A volte viene rilevata macrocitosi; la resistenza osmotica degli eritrociti è ridotta, durante le crisi il contenuto di emoglobina libera nel plasma aumenta, l'emosiderina può comparire nelle urine |
Agenti di disintossicazione, trasfusione di sangue, splenectomia per forma cronica. |
|
Anemia emolitica associata a deficit di enzimi coinvolti nell'utilizzo dell'ATP |
Carenza di adenosina trifosfatasi, adenilato chinasi, ribofosfato-pirofosfato chinasi. La durata della vita dei globuli rossi è ridotta. I globuli rossi vengono emolizzati nella milza |
L'ittero si esprime al momento della crisi emolitica. L’anemia è più pronunciata nella forma acuta che in quella cronica. La milza e il fegato talvolta sono ingrossati |
Crisi emolitiche e aplastiche; calcoli biliari pigmentati |
A volte viene rilevata macrocitosi; la resistenza osmotica degli eritrociti è ridotta, durante le crisi il contenuto di emoglobina libera nel plasma aumenta, l'emosiderina può comparire nelle urine |
|
|
Anemia emolitica associata a deficit di enzimi coinvolti nella sintesi delle porfirine |
Carenza di enzimi coinvolti nella sintesi delle porfirine. La durata della vita dei globuli rossi è ridotta. I globuli rossi vengono emolizzati nella milza |
L'ittero si esprime al momento della crisi emolitica. L’anemia è più pronunciata durante le condizioni croniche. La milza e il fegato talvolta sono ingrossati |
Crisi emolitiche e aplastiche; calcoli biliari pigmentati |
A volte viene rilevata macrocitosi; la resistenza osmotica degli eritrociti è ridotta, durante le crisi il contenuto di emoglobina libera nel plasma aumenta, l'emosiderina può comparire nelle urine |
Agenti disintossicanti, trasfusione di sangue, splenectomia. |
|
Emoglobinopatie |
|||||
|
Emoglobinopatie qualitative |
Violazione della struttura delle catene globiniche con una violazione della sequenza di aminoacidi e formazione di emoglobine anormali. I globuli rossi sono emolizzati nella milza e nel fegato |
Ittero persistente, ingrossamento variabile del fegato e della milza |
Crisi trombotiche; osteomielite; hron, ulcere alle gambe; emosiderosi d'organo |
Eritrociti a forma di falce in presenza di emoglobina S, con altri eritrociti emoglobinosici senza caratteristiche; la resistenza osmotica degli eritrociti è ridotta. Il test diretto di Coombs è negativo; non c'è emosiderina nelle urine, il contenuto di emoglobina libera nel plasma è normale |
Trasfusione di sangue, somministrazione di deferoxamina per emosiderosi, terapia anticoagulante per lo sviluppo di trombosi. |
|
Talassemia |
Sintesi alterata delle catene 0 della globina con la formazione di emoglobina F o A2 o catene alfa con la formazione di emoglobina H o Bart. I globuli rossi vengono emolizzati nel fegato e nella milza |
Vari gradi di ittero costante, la milza è ingrossata e talvolta il fegato è ingrossato; nelle forme gravi, deformazione ossea |
Emosiderosi degli organi, possibile sviluppo di trombosi |
Anemia di vario grado, globuli rossi a forma di bersaglio, diminuzione della resistenza osmotica dei globuli rossi, aumento dell'emoglobina F, A2, H o Bart. Il test diretto di Coombse è negativo |
Trasfusione di sangue, per emosiderosi - somministrazione di deferoxamina. |
|
ANEMIA EMOLITICA ACQUISITA |
|||||
|
Anemie immunoemolitiche |
|||||
|
Anemie emolitiche autoimmuni |
|||||
|
Anemie emolitiche causate da anticorpi caldi |
Aumento della fagocitosi dei globuli rossi principalmente nella milza |
Pallore e ittero costanti, la milza è costantemente ingrossata, il fegato è ingrossato in 1/3 dei pazienti |
Formazione di calcoli biliari; trombosi |
Forte anisocitosi degli eritrociti, diminuzione della loro resistenza osmotica, test diretto di Coombs positivo |
Prednisone in una dose di almeno 1 mg per 1 kg di peso corporeo al giorno; splenectomia |
|
Anemia emolitica causata da anticorpi del raffreddore |
Agglutinazione e morte accelerata dei globuli rossi. |
Pallore e ittero moderati, l'ingrossamento del fegato e della milza non è costante |
Flusso sanguigno alterato nei piccoli vasi |
A volte si osserva una leggera sferocitosi, la resistenza osmotica degli eritrociti è normale, dopo raffreddamento, comparsa di emosiderina nelle urine e aumento del contenuto di emoglobina libera nel plasma |
Immunosoppressori (leukeran) |
|
Anemia emolitica causata da emolisine bifasiche (emoglobinuria parossistica fredda) |
Presenza di emolisina bifasica di Donath-Landsteiner nel sangue. I globuli rossi vengono emolizzati nel letto vascolare |
Stato generale grave, sintomi di mancanza di respiro, febbre. La comparsa di ittero, anemia, emoglobinuria. La milza e il fegato sono moderatamente ingrossati, alquanto dolorosi |
Insufficienza renale, anuria |
Grave anemia normocromica, punteggiatura basofila dei globuli rossi, reticolocitosi, neutrofilia, iperbilirubinemia, aumento dell’emoglobina libera nel plasma |
Misure anti-shock, trasfusione di sangue, somministrazione di poliglucina, diuretici |
|
Anemie immunoemolitiche indotte da farmaci |
Formazione di anticorpi contro il complesso farmaco-eritrociti; talvolta induzione di anticorpi antieritrociti. I globuli rossi vengono emolizzati nel letto vascolare e nella milza |
Pallore e ittero di varia intensità, il fegato e la milza non sono ingranditi |
Emolitico |
La morfologia degli eritrociti è insignificante, la loro resistenza osmotica è normale; al momento della crisi e successivamente, comparsa di emosiderina nelle urine e aumento del contenuto di emoglobina libera nel plasma |
Ritiro urgente del farmaco che ha causato l'emolisi; per anemia grave - trasfusione di sangue |
|
Anemie emolitiche isoimmuni |
|||||
|
Malattia emolitica del neonato |
Isoimmunizzazione materna con antigeni eritrocitari fetali secondo i sistemi Rh, ABO, ecc. I globuli rossi vengono emolizzati principalmente nel fegato e nella milza, parzialmente nel letto vascolare |
Ittero persistente, fegato e milza sono raramente ingranditi |
Bilirubina encefalopatia; rigonfiamento |
Viene spesso rilevata autoagglutinazione degli eritrociti, la resistenza osmotica degli eritrociti è normale, il test diretto di Coombs è negativo, il test indiretto è positivo |
Scambiare trasfusioni di sangue. |
|
Anemia emolitica post-trasfusionale |
Isoimmunizzazione del ricevente con antigeni eritrocitari del donatore (o del feto) con distruzione degli eritrociti trasfusi, meno spesso distruzione degli eritrociti da parte di anticorpi naturali (donatore universale pericoloso). |
Al momento dell'emolisi, pallore pronunciato e ittero, il fegato e la milza non sono ingranditi |
Nefrosi emoglobinurica; insufficienza renale acuta |
Morfologia del sangue senza particolarità, la resistenza osmotica degli eritrociti è normale; il test indiretto di Coombs è positivo, il test diretto è negativo, al momento della crisi comparsa di emosiderina nelle urine e aumento del contenuto di emoglobina libera nel plasma |
Trasfusioni di sangue sostitutive, trattamento dell'insufficienza renale acuta. |
|
Membranopatie acquisite |
|||||
|
Emoglobinuria parossistica notturna |
C'è una carenza di acidi grassi insaturi nella membrana degli eritrociti, che aumenta la sensibilità degli eritrociti al complemento, contribuendo alla loro distruzione. I globuli rossi vengono emolizzati nel letto vascolare |
Pallore grave e ittero moderato, il fegato e la milza di solito non sono ingranditi. Quando si verifica la trombosi, dolore di varie localizzazioni |
Crisi emolitiche e aplastiche, trombosi vascolari |
Anemia grave, diminuzione del numero di leucociti e piastrine; la resistenza osmotica degli eritrociti è ridotta. Il contenuto di emoglobina nel plasma aumenta costantemente, l'emosiderina viene rilevata nelle urine; I test del prosciutto e del saccarosio sono positivi, il test diretto di Coombs è negativo, quello indiretto può essere positivo |
Trasfusione di globuli rossi lavati; somministrazione di ormoni anabolizzanti, con lo sviluppo di trombosi - terapia anticoagulante. Se indicata, splenectomia |
|
Anemia emolitica a cellule speroni |
L'indice colesterolo-fosfolipidi è aumentato nella membrana eritrocitaria; il contenuto di acido litocolico nel plasma è aumentato; la capacità dei globuli rossi di filtrare è ridotta. I globuli rossi vengono emolizzati nella milza |
Ittero, il fegato è raramente ingrossato, la milza è costantemente ingrandita |
Crisi emolitiche e aplastiche; calcoli biliari pigmentati |
Sulla superficie dei globuli rossi sono presenti numerosi piccoli processi simili a spine. Gli altri risultati di laboratorio sono gli stessi dell'anemia emolitica acantocitica |
Splenectomia |
|
Anemia emolitica associata a danno meccanico ai globuli rossi |
|||||
|
Emoglobinuria in marcia |
Le ragioni che causano una maggiore distruzione dei globuli rossi non sono state identificate. Non è stato rilevato alcun danno alla membrana eritrocitaria. Forse la patologia è dovuta alla disposizione insolita dei vasi dei piedi. I globuli rossi vengono emolizzati nel letto vascolare |
La comparsa di urina nera, dolore e malessere nella parte bassa della schiena, debolezza alle gambe dopo aver camminato o corso. A volte lieve ittero e pallore. Il fegato e la milza non sono ingranditi |
Emolitico |
La morfologia del sangue non è cambiata, l'anemia è rara; la resistenza osmotica degli eritrociti è normale; dopo aver camminato, il contenuto di emoglobina libera nel plasma aumenta, l'emosiderina appare nelle urine; i test di Coombs diretti e indiretti sono negativi |
Di solito il trattamento non è necessario. Le misure preventive svolgono un ruolo importante |
|
Malattia di Moschkovich (anemia emolitica sin. microangiopatica) |
Produzione di un complesso antigene-anticorpo quando virus, microbi o vaccini entrano nel sangue; coagulazione intravascolare disseminata, distruzione meccanica dei globuli rossi da parte dei fili di fibrina. I globuli rossi vengono emolizzati nel letto vascolare |
Si sviluppa sullo sfondo di una malattia di base: collagenosi, glomerulonefrite acuta, carcinosi disseminata, porpora trombotica trombocitopenica. Pallore grave e ittero moderato; con lo sviluppo della coagulazione intravascolare, emorragie di varie localizzazioni; il fegato e la milza solitamente non sono ingranditi |
Crisi emolitiche; sintomi di coagulazione intravascolare disseminata (trombosi ed emorragia di varia localizzazione e intensità); fallimento renale cronico |
Globuli rossi globulari e schistociti, anemia grave. Il contenuto di emoglobina libera nel plasma aumenta, l'emosiderina viene rilevata nelle urine; i test di Coombs diretti e indiretti sono negativi. Contenuto ridotto di fattori I, II, VII, VIII e X |
Trattamento della malattia di base; terapia trombolitica; trasfusione di sangue. |
|
Anemia emolitica durante la sostituzione della valvola cardiaca |
Distruzione meccanica dei globuli rossi o rottura della loro membrana. I globuli rossi vengono emolizzati nel letto vascolare |
Pallore e ittero moderati, i bordi si intensificano con il movimento attivo del paziente; il fegato e la milza non sono ingrossati |
Non descritto |
La morfologia del sangue non è cambiata, l'anemia moderata, la resistenza osmotica degli eritrociti è normale; i test di Coombs diretti e indiretti sono negativi. Il contenuto di emoglobina libera nel plasma aumenta, l'emosiderina appare nelle urine |
Nei casi più gravi, intervento chirurgico di ricostruzione della valvola; trasfusione di sangue |
|
Anemia emolitica tossica |
|||||
|
Anemia emolitica tossica di varie eziologie |
Il meccanismo dell'emolisi è diverso. I globuli rossi vengono emolizzati principalmente nel letto vascolare |
Pallore e ittero si esprimono al momento della crisi, il fegato e la milza non sono ingranditi |
Non descritto |
La morfologia del sangue non è cambiata, l'anemia si esprime al momento della crisi; la resistenza osmotica degli eritrociti è normale; al momento della crisi, il contenuto di emoglobina libera nel plasma aumenta, l'emosiderina appare nelle urine; i test di Coombs diretti e indiretti sono negativi |
Evitare il contatto con un agente tossico. Trasfusione di sangue |
Bibliografia: Grozdov D. M. e Pa-ts e su p e M. D. Chirurgia delle malattie del sistema sanguigno, M., 1962, bibliogr.; Dosse J. Immunoematologia, trad. da French, M., 1959; Dygin V.P. Malattie autoimmuni sistemi sanguigni, L., 1964, bibliogr.; Idelson L. I., Didkovskiy N.A. e Ermil-ch e N a circa G.V. Anemia emolitica, M., 1975, bibliogr.; Kassirsky I. A. e Alekseev G. A. Ematologia clinica, M., 1970; Lorie Yu I. Classificazione delle anemie emolitiche, Problemi, ematolo. e traboccare. sangue, vol.7, JVe 9, p. 3, 1962; Boorman K.E., Dodd V.E.a. L o u-tit J. F. Ittero emolitico (ittero acholurico), Lancet, v. 1, pag. 812, 1946; Brain M. C. I globuli rossi e l'anemia emolitica, nel libro: Recent advanc. haema-tol., ed. di A. Goldberg a. M. G. Brain, pag. 146, Edimburgo - L., 1971, bibliogr.; CervelloM. G., Dacie J. V. a. H o u r i-h a n e D. O. Anemia emolitica microangiopatica, Brit. J. Haemat., y. 8, pag. 358, 1962; Ghauffard A. L'ictfcre h6moly-tique congenital de 1'adulte, Rev. g£n. Clinica. Là., t. 26, pag. 199, 1912; D ac i e J. Y. Le anemie emolitiche, pt 1-4, L., 1962-1967; Gerbal A. e. UN. Nouvelle classification immunologique des anemies hemolytiques avec auto-anticorps, Nouv. Rev. frang, H&nat., t. 7, pag. 401, 1967; H a r r i s J. W. Studi sul meccanismo dell'anemia emolitica indotta da farmaci, J. Lab. clin. Med., v. 44, pag. 809, 1954; Hennemann H. H. Erworbene hamo-lytische Anamien, Lpz., 1957, Bibliogr.; Libanskt J. Storungen der Erythropoese medikamentosen Ursprungs, nel libro: Fortschr. Hamatol., hrsg. v. E. Perlick u. a., Bd 1, S. 125, Lpz., 1970, Bibliogr.; Williams WJ a. o. Ematologia, New York, 1972.
Anatomia patologica- Ageev A.K. Caratteristiche morfologiche delle anemie emolitiche autoimmuni che si sviluppano nella leucemia, Arch. pathol., t.26, n.1, p. 71, 1964; Danilova L. A. Cambiamenti patologici nella milza nell'anemia emolitica, Problemi, ematolo. e traboccamento, sangue, vol.5, n.7, p. 19, 1960; alias, Aspetti patologici delle forme congenite e acquisite di anemia emolitica cronica, arch. pathol., t.27, n. I, p. 3, 1965; A n d r 6 R. e. UN. Anomie h6molytiques autoimmuni avec noduli linfoidi della moella osseuse, Sem. Salto. Parigi, pag. 2517, 1968; Con o line n G. u. UN. Tumorartige intrathorakale extramedullare Hamatopoese bei hamoly-tischer Anamie, Acta haemat. (Basilea), y. 43, pag. 111, 1970; LeonardiP. UN. RuolA. Emosiderosi renale nelle anemie emolitiche, diagnosi mediante agobiopsia, Sangue, v. 16, pag. 1029, 1960; M o t u 1 s k y A. G., G r o s by W. H. a. Rappaport H. Malattia emolitica ereditaria non sferocitica, ibid., v. 9, pag. 749, 1954; Neumark E. Apparizioni post mortem nell'anemia emolitica, Sang, p. 161, 1950; Pirofsky B. Autoimmunizzazione e anemie emolitiche autoimmuni, Baltimora, 1969; Rapporto rap H. a. Crosby W. H. Anemia emolitica autoimmune, Osservazioni morfologiche e correlazioni clinicopatologiche, Amer. J* Sentiero., v. 33, pag. 429, 1957.
Yu.I. Lorie; G. A. Alekseev (emoglobinuria), M. P. Khokhlova (pat. an.), compilatore della tabella Yu. I. Lorie.
Che sono caratterizzati da una diminuzione della durata della vita dei globuli rossi e dalla loro distruzione accelerata (emolisi, eritrocitolisi) sia all'interno dei vasi sanguigni che nel midollo osseo, nel fegato o nella milza.
Ciclo vitale globuli rossi nell'anemia emolitica – 15-20 giorni
Bene durata media La durata della vita degli eritrociti è di 110-120 giorni. Nell'anemia emolitica, il ciclo di vita dei globuli rossi si riduce più volte e ammonta a 15-20 giorni. I processi di distruzione dei globuli rossi prevalgono sui processi della loro maturazione (eritropoiesi), a seguito dei quali diminuisce la concentrazione di emoglobina nel sangue, diminuisce il contenuto dei globuli rossi, cioè si sviluppa l'anemia. Altri caratteristiche comuni, caratteristici di tutti i tipi di anemia emolitica, sono:
- febbre con brividi;
- dolore all'addome e alla parte bassa della schiena;
- disturbi del microcircolo;
- splenomegalia (milza ingrossata);
- emoglobinuria (presenza di emoglobina nelle urine);
L’anemia emolitica colpisce circa l’1% della popolazione. Nell'assetto complessivo delle anemie, quelle emolitiche rappresentano l'11%.
Cause dell'anemia emolitica e fattori di rischio
Le anemie emolitiche si sviluppano sotto l'influenza di fattori extracellulari (esterni) o come risultato di difetti dei globuli rossi (fattori intracellulari). Nella maggior parte dei casi, i fattori extracellulari vengono acquisiti mentre i fattori intracellulari sono congeniti.
 I difetti dei globuli rossi sono un fattore intracellulare nello sviluppo dell'anemia emolitica
I difetti dei globuli rossi sono un fattore intracellulare nello sviluppo dell'anemia emolitica
I fattori intracellulari comprendono anomalie delle membrane dei globuli rossi, degli enzimi o dell'emoglobina. Tutti questi difetti sono ereditari, ad eccezione dell'emoglobinuria parossistica notturna. Attualmente sono state descritte oltre 300 malattie associate a mutazioni puntiformi di geni che codificano per la sintesi delle globine. Come risultato delle mutazioni, la forma e la membrana dei globuli rossi cambiano e aumenta la loro suscettibilità all'emolisi.
Un gruppo più ampio è rappresentato dai fattori extracellulari. I globuli rossi sono circondati dall'endotelio (il rivestimento interno) dei vasi sanguigni e dal plasma. La presenza di agenti infettivi, sostanze tossiche e anticorpi nel plasma può causare cambiamenti nelle pareti dei globuli rossi, portando alla loro distruzione. Con questo meccanismo, ad esempio, si sviluppano l'anemia emolitica autoimmune e le reazioni trasfusionali emolitiche.
Difetti nell'endotelio dei vasi sanguigni (microangiopatia) possono anche danneggiare i globuli rossi, portando allo sviluppo dell'anemia emolitica microangiopatica, che si manifesta acutamente nei bambini sotto forma di sindrome emolitico-uremica.
L'anemia emolitica può essere causata anche dall'assunzione di alcuni farmaci, in particolare antimalarici, analgesici, nitrofurani e sulfamidici.
Fattori provocatori:
- vaccinazione;
- malattie autoimmuni (colite ulcerosa, lupus eritematoso sistemico);
- alcune malattie infettive (polmonite virale, sifilide, toxoplasmosi, mononucleosi infettiva);
- enzimopatie;
- emoblastosi (mieloma, linfogranulomatosi, leucemia linfocitica cronica, leucemia acuta);
- avvelenamento con arsenico e suoi composti, alcool, funghi velenosi, acido acetico, metalli pesanti;
- pesante esercizio fisico(sci lungo, corsa o camminata per lunghe distanze);
- ipertensione arteriosa maligna;
- bruciare la malattia;
- protesi di vasi sanguigni e valvole cardiache.
Forme della malattia
Tutte le anemie emolitiche si dividono in acquisite e congenite. Le forme congenite o ereditarie includono:
- membranopatie eritrocitarie– il risultato di anomalie nella struttura delle membrane degli eritrociti (acantocitosi, ovalocitosi, microsferocitosi);
- enzimopenia (fermentopenia)– associato a una carenza di alcuni enzimi nell’organismo (piruvato chinasi, glucosio-6-fosfato deidrogenasi);
- emoglobinopatie– causato da una violazione della struttura della molecola di emoglobina ( anemia falciforme, talassemia).
L’anemia emolitica ereditaria più comune nella pratica clinica è la malattia di Minkowski-Choffard (microsferocitosi).
Le anemie emolitiche acquisite, a seconda delle cause che le hanno provocate, si suddividono nelle seguenti tipologie:
- membranopatie acquisite(anemia a cellule sperone, emoglobinuria parossistica notturna);
- Anemie emolitiche isoimmuni e autoimmuni– si sviluppano a seguito del danno ai globuli rossi da parte degli anticorpi propri o ricevuti esternamente;
- tossico– la distruzione accelerata dei globuli rossi avviene a causa dell’esposizione a tossine batteriche, veleni biologici o sostanze chimiche;
- anemia emolitica associata a danno meccanico ai globuli rossi(emoglobinuria in marcia, porpora trombocitopenica).
Tutti i tipi di anemia emolitica sono caratterizzati da:
- sindrome anemica;
- milza ingrossata;
- sviluppo di ittero.
Inoltre, ogni singolo tipo di malattia ha le sue caratteristiche.

Anemie emolitiche ereditarie
L’anemia emolitica ereditaria più comune nella pratica clinica è la malattia di Minkowski-Choffard (microsferocitosi). È rintracciabile in diverse generazioni della famiglia ed è ereditata con modalità autosomica dominante. Una mutazione genetica porta a livelli insufficienti di alcuni tipi di proteine e lipidi nella membrana dei globuli rossi. A sua volta, ciò provoca cambiamenti nella dimensione e nella forma dei globuli rossi, la loro prematura distruzione massiccia nella milza. L'anemia emolitica microsferocitica può manifestarsi in pazienti di qualsiasi età, ma molto spesso i primi sintomi dell'anemia emolitica compaiono tra i 10 e i 16 anni.
 La microsferocitosi è l’anemia emolitica ereditaria più comune
La microsferocitosi è l’anemia emolitica ereditaria più comune
La malattia può manifestarsi con gravità variabile. Alcuni pazienti presentano un decorso subclinico, mentre altri sviluppano forme gravi, accompagnate da frequenti crisi emolitiche, che presentano le seguenti manifestazioni:
- aumento della temperatura corporea;
- brividi;
- debolezza generale;
- dolore alla parte bassa della schiena e all'addome;
- nausea.
Il sintomo principale della microsferocitosi è vari gradi gravità dell'ittero. A causa dell'alto contenuto di stercobilina (il prodotto finale del metabolismo dell'eme), le feci hanno un colore marrone scuro intenso. In tutti i pazienti affetti da anemia emolitica microsferocitica, la milza è ingrandita e in ogni secondo paziente il fegato è ingrandito.
La microsferocitosi aumenta il rischio di formazione di calcoli nella cistifellea, cioè lo sviluppo di colelitiasi. A questo proposito, spesso si verifica una colica biliare e quando il dotto biliare è bloccato da un calcolo si verifica un ittero ostruttivo (meccanico).
Il quadro clinico dell'anemia emolitica microsferocitica nei bambini contiene anche altri segni di displasia:
- Bradidattilia o polidattilia;
- cielo gotico;
- malocclusione;
- deformità del naso a sella;
- teschio della torre.
Nei pazienti anziani, a causa della distruzione dei globuli rossi nei capillari degli arti inferiori, si verificano ulcere trofiche dei piedi e delle gambe, resistenti alla terapia tradizionale.
L'anemia emolitica, associata a una carenza di alcuni enzimi, si manifesta solitamente dopo l'assunzione di determinati farmaci o dopo aver sofferto di una malattia intercorrente. I loro tratti caratteristici sono:
- ittero pallido ( colore pallido pelle con tinta limone);
- soffi al cuore;
- epatosplenomegalia moderatamente grave;
- colore scuro delle urine (dovuto alla rottura intravascolare dei globuli rossi e al rilascio di emosiderina nelle urine).
Nei casi più gravi della malattia si verificano crisi emolitiche pronunciate.
Le emoglobinopatie congenite comprendono la talassemia e l'anemia falciforme. Il quadro clinico della talassemia è espresso dai seguenti sintomi:
- anemia ipocromica;
- emocromatosi secondaria (associata a frequenti trasfusioni di sangue e prescrizione irragionevole di farmaci contenenti ferro);
- ittero emolitico;
- splenomegalia;
- colelitiasi;
- danno articolare (artrite, sinovite).
L'anemia falciforme si manifesta con crisi dolorose ricorrenti, moderata anemia emolitica e maggiore suscettibilità del paziente alle malattie infettive. I sintomi principali sono:
- ritardo dei bambini nello sviluppo fisico (soprattutto ragazzi);
- ulcere trofiche degli arti inferiori;
- ittero moderato;
- crisi dolorose;
- crisi aplastiche ed emolitiche;
- priapismo (erezione spontanea del pene, non associata all'eccitazione sessuale, della durata di diverse ore);
- colelitiasi;
- splenomegalia;
- necrosi avascolare;
- osteonecrosi con sviluppo di osteomielite.

Anemia emolitica acquisita
Tra le anemie emolitiche acquisite, le più comuni sono quelle autoimmuni. Il loro sviluppo è causato dalla produzione di anticorpi da parte del sistema immunitario del paziente diretti contro i propri globuli rossi. Cioè, sotto l'influenza di alcuni fattori, l'attività del sistema immunitario viene interrotta, a seguito della quale inizia a percepire i propri tessuti come estranei e a distruggerli.
A anemia autoimmune le crisi emolitiche si sviluppano improvvisamente e acutamente. La loro comparsa può essere preceduta da precursori sotto forma di artralgia e/o febbre bassa corpi. I sintomi di una crisi emolitica sono:
- aumento della temperatura corporea;
- vertigini;
- grave debolezza;
- dispnea;
- battito cardiaco;
- dolore alla parte bassa della schiena e all'epigastrio;
- rapido aumento dell'ittero, non accompagnato da prurito della pelle;
- ingrossamento della milza e del fegato.
Esistono forme di anemia emolitica autoimmune in cui i pazienti non tollerano bene il freddo. Quando si verifica l'ipotermia, sviluppano emoglobinuria, orticaria da freddo e sindrome di Raynaud (grave spasmo delle arteriole delle dita).
Le caratteristiche del quadro clinico delle forme tossiche di anemia emolitica sono:
- debolezza generale rapidamente progressiva;
- temperatura corporea elevata;
- vomito;
- forte dolore alla parte bassa della schiena e all'addome;
- emoglobinuria.
Entro 2-3 giorni dall'esordio della malattia, il livello di bilirubina nel sangue del paziente inizia ad aumentare e si sviluppa l'ittero, e dopo altri 1-2 giorni si verifica un'insufficienza epatorenale, manifestata da anuria, azotemia, fermentemia, epatomegalia.
Un'altra forma di anemia emolitica acquisita è l'emoglobinuria. Con questa patologia, si verifica una massiccia distruzione dei globuli rossi all'interno dei vasi sanguigni e l'emoglobina entra nel plasma e quindi inizia ad essere escreta nelle urine. Il sintomo principale dell'emoglobinuria è il colore rosso scuro (a volte nero) delle urine. Altre manifestazioni di patologia possono includere:
- Forte mal di testa;
- un forte aumento della temperatura corporea;
- brividi tremendi;
L'emolisi degli eritrociti nella malattia emolitica del feto e dei neonati è associata alla penetrazione di anticorpi dal sangue materno nel flusso sanguigno fetale attraverso la placenta, ad es. meccanismo patologico Questa forma di anemia emolitica è una malattia isoimmune.
Normalmente, la durata media della vita dei globuli rossi è di 110-120 giorni. Nell'anemia emolitica, il ciclo di vita dei globuli rossi si riduce più volte e ammonta a 15-20 giorni.
La malattia emolitica del feto e del neonato può manifestarsi in uno dei seguenti modi:
- morte fetale intrauterina;
- forma edematosa (forma immune di idrope fetale);
- forma itterica;
- forma anemica.
I sintomi comuni caratteristici di tutte le forme di questa malattia sono:
- epatomegalia;
- splenomegalia;
- un aumento degli eritroblasti nel sangue;
- anemia normocromica.
Diagnostica
I pazienti con anemia emolitica vengono esaminati da un ematologo. Durante l'intervista al paziente, scoprono la frequenza delle crisi emolitiche, la loro gravità e chiariscono anche la presenza di malattie simili nella storia familiare. Durante l'esame del paziente si presta attenzione al colore della sclera, delle mucose visibili e della pelle e si palpa l'addome per identificare possibili ingrossamenti del fegato e della milza. L'ecografia degli organi addominali può confermare l'epatosplenomegalia.
I cambiamenti nell'esame del sangue generale con anemia emolitica sono caratterizzati da anemia ipo o normocromica, reticolocitosi, trombocitopenia, emoglobinuria, emosiderinuria, urobilinuria, proteinuria. C'è un aumento del contenuto di stercobilina nelle feci.
Se necessario, viene eseguita una biopsia puntura del midollo osseo, seguita da analisi istologica(viene rilevata l'iperplasia della linea eritroide).
L’anemia emolitica colpisce circa l’1% della popolazione. Nell'assetto complessivo delle anemie, quelle emolitiche rappresentano l'11%.
La diagnosi differenziale dell'anemia emolitica viene effettuata con le seguenti malattie:
- emoblastosi;
- sindrome epatolienale;
- ipertensione portale;
- cirrosi epatica;
Trattamento delle anemie emolitiche
Gli approcci al trattamento dell'anemia emolitica sono determinati dalla forma della malattia. Ma in ogni caso, il compito principale è eliminare il fattore emolizzante.
Regime di trattamento per la crisi emolitica:
- infusione endovenosa di soluzioni elettrolitiche e di glucosio;
- trasfusione di plasma sanguigno fresco congelato;
- terapia vitaminica;
- prescrizione di antibiotici e/o corticosteroidi (come indicato).

Per la microsferocitosi è indicato il trattamento chirurgico: rimozione della milza (splenectomia). Dopo l'intervento chirurgico, il 100% dei pazienti sperimenta remissione stabile, poiché l'aumento dell'emolisi dei globuli rossi si interrompe.
La terapia per l'anemia emolitica autoimmune viene effettuata con ormoni glucocorticoidi. Se non è sufficientemente efficace, potrebbe essere necessario prescrivere immunosoppressori, farmaci antimalarici. Resistenza terapia farmacologicaè un'indicazione alla splenectomia.
Per l'emoglobinuria, vengono effettuate trasfusioni di globuli rossi lavati, infusione di soluzioni di espansori plasmatici e vengono prescritti agenti antipiastrinici e anticoagulanti.
Il trattamento delle forme tossiche di anemia emolitica richiede l'introduzione di antidoti (se disponibili), nonché l'uso di metodi di disintossicazione extracorporea (diuresi forzata, dialisi peritoneale, emodialisi, emosorbimento).
Possibili conseguenze e complicazioni
L'anemia emolitica può portare allo sviluppo delle seguenti complicanze:
- attacchi di cuore e rottura della milza;
- sindrome DIC;
- Coma emolitico (anemico).
Previsione
Con un trattamento tempestivo e adeguato dell'anemia emolitica, la prognosi è generalmente favorevole. Quando sorgono complicazioni, peggiora in modo significativo.
Prevenzione
La prevenzione dello sviluppo dell'anemia emolitica comprende le seguenti misure:
- consulenza medica e genetica alle coppie se esiste una storia familiare indicante casi di anemia emolitica;
- determinazione del gruppo sanguigno e del fattore Rh della futura mamma nella fase di pianificazione della gravidanza;
- rafforzare il sistema immunitario.
Video da YouTube sull'argomento dell'articolo:
L'anemia emolitica si riferisce a malattie caratterizzate come anemia. La forma ereditaria più comune della malattia, che può svilupparsi anche nei neonati. In generale, questa malattia del sangue può essere riscontrata in persone di qualsiasi età (e anche negli animali domestici, come i cani) e ha eziologie sia congenite che acquisite. A causa della sua capacità dannosa, l'anemia emolitica è una malattia molto pericolosa ed è difficile da curare, richiede molto tempo e in ambito ospedaliero.
Particolarmente pericolosa è la crisi emolitica, che provoca forte peggioramento le condizioni del paziente e richiedono un intervento urgente. Le forme avanzate della malattia portano all'intervento chirurgico, il che indica la necessità di un rilevamento tempestivo e di un trattamento efficace.
L'essenza della malattia
L'anemia emolitica comprende un intero gruppo di anemie con aumento dell'emolisi degli eritrociti, aumento dei livelli di prodotti di distruzione degli eritrociti in combinazione con la presenza di eritropoiesi potenziata in modo reattivo. L'essenza della malattia è l'aumento della distruzione del sangue dovuta a una significativa riduzione del ciclo di vita dei globuli rossi a causa del fatto che la loro distruzione avviene più velocemente della formazione di nuovi.
 Gli eritrociti (globuli rossi) sono cellule del sangue responsabili di molti funzione importante- trasferimento di ossigeno agli organi interni. Si formano nel midollo osseo rosso e dopo la maturazione entrano nel flusso sanguigno e si diffondono con esso in tutto il corpo. Il ciclo vitale di queste cellule è di circa 100-120 giorni, la loro morte giornaliera raggiunge l'1% del numero totale. È questa quantità che viene sostituita da una nuova, che mantiene il livello normale di globuli rossi nel sangue.
Gli eritrociti (globuli rossi) sono cellule del sangue responsabili di molti funzione importante- trasferimento di ossigeno agli organi interni. Si formano nel midollo osseo rosso e dopo la maturazione entrano nel flusso sanguigno e si diffondono con esso in tutto il corpo. Il ciclo vitale di queste cellule è di circa 100-120 giorni, la loro morte giornaliera raggiunge l'1% del numero totale. È questa quantità che viene sostituita da una nuova, che mantiene il livello normale di globuli rossi nel sangue.
Come risultato della patologia nei vasi periferici o nella milza, si verifica una distruzione accelerata dei globuli rossi e le nuove cellule non hanno il tempo di svilupparsi: il loro equilibrio nel sangue viene interrotto. Di riflesso, il corpo attiva la formazione di globuli rossi nel midollo osseo, ma non hanno il tempo di maturare e i giovani globuli rossi immaturi - i reticolociti - entrano nel sangue, causando il processo di emolisi.
Patogenesi della malattia
La patogenesi dell'anemia emolitica si basa sulla distruzione dei globuli rossi con diffusione dell'emoglobina e formazione di bilirubina. Il processo di distruzione dei globuli rossi può avvenire in due modi: intracellulare e intravascolare.
 L'emolisi intracellulare o extravascolare si sviluppa nei macrofagi della milza, meno spesso nel midollo osseo e nel fegato. Processo distruttivo causato dalla patologia della membrana eritrocitaria o dalla limitazione della loro capacità di cambiare forma, causata dall'inferiorità morfologica e funzionale congenita di queste cellule. Nel sangue c'è un aumento significativo della concentrazione di bilirubina e una diminuzione del contenuto di aptoglobina. I principali rappresentanti di questa variante della patogenesi sono le anemie emolitiche autoimmuni.
L'emolisi intracellulare o extravascolare si sviluppa nei macrofagi della milza, meno spesso nel midollo osseo e nel fegato. Processo distruttivo causato dalla patologia della membrana eritrocitaria o dalla limitazione della loro capacità di cambiare forma, causata dall'inferiorità morfologica e funzionale congenita di queste cellule. Nel sangue c'è un aumento significativo della concentrazione di bilirubina e una diminuzione del contenuto di aptoglobina. I principali rappresentanti di questa variante della patogenesi sono le anemie emolitiche autoimmuni.
L'emolisi intravascolare avviene direttamente nei canali del sangue sotto l'influenza di fattori esterni, come lesioni meccaniche, lesioni tossiche, trasfusione di sangue incompatibile, ecc. La patologia è accompagnata dal rilascio di emoglobina libera nel plasma sanguigno e dall'emoglobinuria. Come risultato della formazione di metaemoglobina, il siero del sangue diventa marrone e il livello di aptoglobina diminuisce drasticamente. L’emoglobinuria può causare insufficienza renale.
Entrambi i meccanismi di patogenesi sono pericolosi a causa della loro manifestazione estrema: una crisi emolitica, quando l'emolisi degli eritrociti diventa diffusa, che porta ad una forte progressione dell'anemia e ad un deterioramento delle condizioni di una persona.
Classificazione della malattia
 Tenendo conto dell'eziologia della malattia, l'anemia emolitica è divisa in due tipi principali: acquisita e congenita; Anche in medicina si distinguono specie specifiche più rare. Le anemie emolitiche congenite o ereditarie comprendono le seguenti forme principali della malattia:
Tenendo conto dell'eziologia della malattia, l'anemia emolitica è divisa in due tipi principali: acquisita e congenita; Anche in medicina si distinguono specie specifiche più rare. Le anemie emolitiche congenite o ereditarie comprendono le seguenti forme principali della malattia:
- Membranopatia: anemia causata da difetti nella struttura dei globuli rossi. Varietà microsferocitiche, ovalocitiche e acantocitiche.
- Forma enzimatica: la patologia è causata da una carenza di vari enzimi: la classe dei pentoso fosfati, la glicolisi, l'ossidazione o la riduzione del glutatione, l'ATP, la sintesi della porfirina.
- Emoglobinopatie: malattie associate ad alterata sintesi di emoglobina, varietà e talassemia.
L'anemia emolitica acquisita presenta le seguenti forme caratteristiche:
- Tipo immunoemolitico: anemia autoimmune isoimmune ed emolitica.
- Membranopatie acquisite: emoglobinuria parossistica notturna e di tipo speronato.
- Basato sul danno meccanico alle cellule: emoglobinuria di tipo microangiopatico (malattia di Moshkovich) e anemia a seguito dell'installazione di valvole cardiache protesiche.
- Varietà tossiche: il tipo principale è l'anemia da farmaci e l'esposizione al veleno emolitico.
 Oltre alle due forme principali della malattia, si distinguono tipi specifici di patologia.
Oltre alle due forme principali della malattia, si distinguono tipi specifici di patologia.
In particolare, l'anemia emolitica nei bambini può esprimersi come ittero emolitico dei neonati.
In questo caso, il danno ai globuli rossi è causato dagli effetti distruttivi degli anticorpi materni.
Un tipo comune di malattia è l'anemia idiopatica, inclusa la forma secondaria causata dal linfoma.
Cause dell'anemia emolitica
Le cause della patologia possono essere suddivise in esterne ed interne, a seconda dei fattori che influenzano. L'anemia congenita è generata da cause interne a livello genetico: eredità di un gene anomalo da uno o entrambi i genitori; manifestazione di spontaneità mutazione genetica durante lo sviluppo del feto nel grembo materno.
La patologia più pericolosa è la forma omozigote, quando il gene anomalo è presente su entrambi i cromosomi della stessa coppia. Tali anemie emolitiche nei bambini hanno una prognosi estremamente pessimistica per lo sviluppo.
 Nell'eziologia delle malattie acquisite si possono distinguere le seguenti ragioni principali: ingestione di veleni (arsenico, veleno di serpente, funghi velenosi, piombo, ecc.); ipersensibilità a determinati prodotti chimici e farmaci; lesione infettiva(malaria, epatite, herpes, infezioni alimentari, ecc.); ustioni; trasfusione di sangue incompatibile per gruppo e fattore Rh; fallimento dentro sistema immunitario, che porta alla produzione di anticorpi contro i propri globuli rossi; danno meccanico ai globuli rossi (esposizione chirurgica); mancanza di vitamina E; assunzione eccessiva di alcuni farmaci (antibiotici, sulfamidici); malattie sistemiche tessuti connettivi (lupus eritematoso, artrite reumatoide).
Nell'eziologia delle malattie acquisite si possono distinguere le seguenti ragioni principali: ingestione di veleni (arsenico, veleno di serpente, funghi velenosi, piombo, ecc.); ipersensibilità a determinati prodotti chimici e farmaci; lesione infettiva(malaria, epatite, herpes, infezioni alimentari, ecc.); ustioni; trasfusione di sangue incompatibile per gruppo e fattore Rh; fallimento dentro sistema immunitario, che porta alla produzione di anticorpi contro i propri globuli rossi; danno meccanico ai globuli rossi (esposizione chirurgica); mancanza di vitamina E; assunzione eccessiva di alcuni farmaci (antibiotici, sulfamidici); malattie sistemiche tessuti connettivi (lupus eritematoso, artrite reumatoide).
Sintomi della malattia
Indipendentemente dal tipo di malattia, si possono identificare sintomi generali caratteristici dell'anemia emolitica: pallore o giallo della pelle, della mucosa orale, degli occhi; tachicardia, debolezza e mancanza di respiro, ingrossamento della milza e del fegato, segni di ittero, vertigini, febbre, possibile annebbiamento della coscienza e convulsioni; un aumento della viscosità del sangue, che porta a trombosi e compromissione dell’afflusso di sangue.
 A seconda del meccanismo della patogenesi, si notano sintomi specifici.
A seconda del meccanismo della patogenesi, si notano sintomi specifici.
Con l'emolisi intravascolare compaiono i seguenti segni: aumento della temperatura, cambiamento del colore delle urine (rosso, marrone o annerito), rilevamento della bilirubina nel sangue, indice di colore compreso tra 0,8 e 1,1.
Il meccanismo intracellulare porta a sintomi come ingiallimento della pelle e delle mucose, diminuzione del livello di emoglobina e globuli rossi nel sangue, numero di reticolociti superiore al 2%, aumento del contenuto di bilirubina indiretta, un grande quantità di urobilina nelle urine e stercobilina nelle feci.
Forme cliniche comuni
Nelle manifestazioni cliniche si possono distinguere diverse forme comuni di anemia emolitica:

- L'anemia di Minkowski-Choffard (microsferocitosi ereditaria) è caratterizzata da un'anomala permeabilità della membrana eritrocitaria attraverso la quale passano gli ioni sodio. La malattia ha carattere ereditario autosomico dominante. Lo sviluppo è ondulato: alternanza di periodi stabili e crisi emolitiche. Segni principali: diminuzione della resistenza osmotica degli eritrociti, predominanza degli eritrociti alterati - microsferociti, reticolocitosi. In caso di un decorso complesso della malattia, è necessario Intervento chirurgico(splenectomia).
- La talassemia comprende una serie di malattie di natura simile che hanno una base ereditaria. La malattia è associata ad una ridotta produzione di emoglobina. Segni principali: milza ingrossata, labbro leporino, cranio a torre, indice cromatico ipocromico, forma alterata dei globuli rossi, reticolocitosi, aumento dei livelli di bilirubina e ferro nel sangue. Quando viene rilevata tale anemia emolitica, il trattamento viene effettuato somministrando globuli rossi e acido folico.
- L’anemia falciforme è il tipo più comune di emoglobinopatia. Segno caratteristico: I globuli rossi assumono la forma di una falce, che li fa rimanere intrappolati nei capillari, provocando la trombosi. Le crisi emolitiche sono accompagnate dal rilascio di urina nera con tracce di sangue, da una significativa diminuzione dell'emoglobina nel sangue e da febbre. Il midollo osseo contiene un alto contenuto di eritrocariociti. Durante il trattamento, al paziente viene somministrato importo aumentato liquidi, viene eseguita l'ossigenoterapia e vengono prescritti antibiotici.
- Si riferiscono alle porfirie forma ereditaria malattie e sono causate da una violazione della formazione di porfirine - componenti dell'emoglobina. Il primo segno è l'ipocromia, appare gradualmente la deposizione di ferro, la forma dei globuli rossi cambia e nel midollo osseo compaiono sideroblasti. La porfiria può anche essere contratta a causa di avvelenamento tossico. Il trattamento viene effettuato somministrando glucosio ed ematite.
- L'anemia emolitica autoimmune è caratterizzata dalla distruzione dei globuli rossi da parte degli anticorpi diretti contro la loro membrana e i linfociti. Il trattamento prevale con l'uso di ormoni steroidei (prednisolone, desametasone e citostatici). Se necessario, viene eseguito il trattamento chirurgico: splenectomia.